Roybal Kole T, Buck Taráz E, Ruan Xiongtao, Cho Baek Hwan, Clark Danielle J, Ambler Rachel, Tunbridge Helen M, Zhang Jianwei, Verkade Paul, Wülfing Christoph, Murphy Robert F
School of Cellular and Molecular Medicine, University of Bristol, Bristol BS8 1TD, UK. Department of Immunology, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Computational Biology Department, School of Computer Science, Carnegie Mellon University, Pittsburgh, PA 15213, USA.
Sci Signal. 2016 Apr 19;9(424):rs3. doi: 10.1126/scisignal.aad4149.
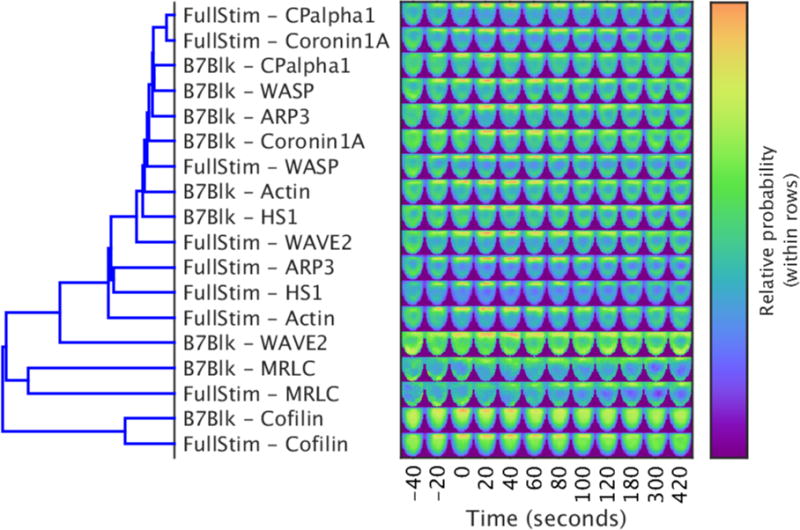
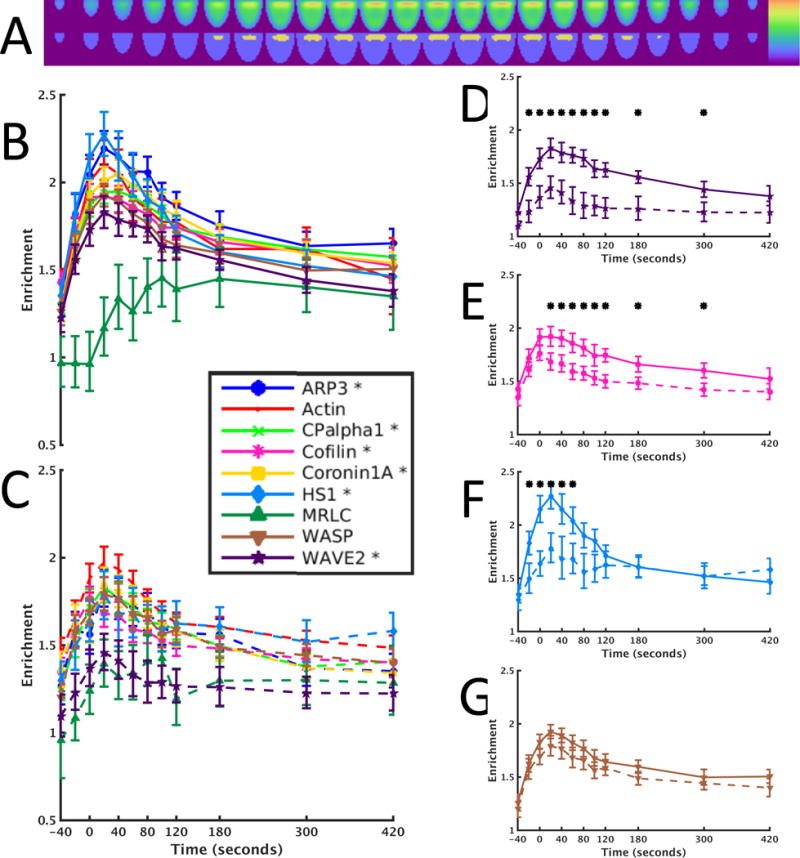
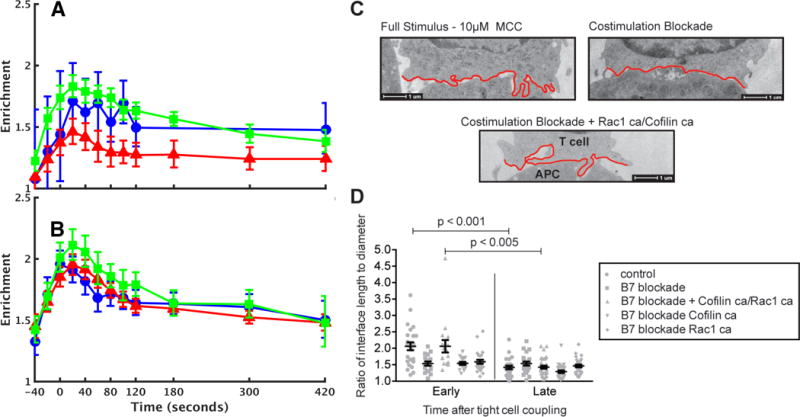
Fluorescence microscopy is one of the most important tools in cell biology research because it provides spatial and temporal information to investigate regulatory systems inside cells. This technique can generate data in the form of signal intensities at thousands of positions resolved inside individual live cells. However, given extensive cell-to-cell variation, these data cannot be readily assembled into three- or four-dimensional maps of protein concentration that can be compared across different cells and conditions. We have developed a method to enable comparison of imaging data from many cells and applied it to investigate actin dynamics in T cell activation. Antigen recognition in T cells by the T cell receptor (TCR) is amplified by engagement of the costimulatory receptor CD28. We imaged actin and eight core actin regulators to generate over a thousand movies of T cells under conditions in which CD28 was either engaged or blocked in the context of a strong TCR signal. Our computational analysis showed that the primary effect of costimulation blockade was to decrease recruitment of the activator of actin nucleation WAVE2 (Wiskott-Aldrich syndrome protein family verprolin-homologous protein 2) and the actin-severing protein cofilin to F-actin. Reconstitution of WAVE2 and cofilin activity restored the defect in actin signaling dynamics caused by costimulation blockade. Thus, we have developed and validated an approach to quantify protein distributions in time and space for the analysis of complex regulatory systems.
荧光显微镜是细胞生物学研究中最重要的工具之一,因为它能提供空间和时间信息来研究细胞内的调节系统。这项技术可以在单个活细胞内数千个分辨位置上以信号强度的形式生成数据。然而,鉴于细胞间存在广泛的差异,这些数据无法轻易组装成可在不同细胞和条件下进行比较的蛋白质浓度三维或四维图谱。我们开发了一种方法,能够比较来自多个细胞的成像数据,并将其应用于研究T细胞活化过程中的肌动蛋白动力学。T细胞受体(TCR)介导的T细胞对抗原的识别通过共刺激受体CD28的结合而得到增强。我们对肌动蛋白和八种核心肌动蛋白调节因子进行成像,以生成一千多部T细胞的电影,这些电影记录了在强TCR信号背景下CD28被结合或阻断的条件下T细胞的情况。我们的计算分析表明,共刺激阻断的主要作用是减少肌动蛋白成核激活剂WAVE2(威斯科特-奥尔德里奇综合征蛋白家族维普洛林同源蛋白2)和肌动蛋白切割蛋白丝切蛋白向F-肌动蛋白的募集。恢复WAVE2和丝切蛋白的活性可修复由共刺激阻断引起的肌动蛋白信号动力学缺陷。因此,我们开发并验证了一种在时间和空间上量化蛋白质分布的方法,用于分析复杂的调节系统。