Kim Byung-Jin, Silverman Sean M, Liu Yang, Wordinger Robert J, Pang Iok-Hou, Clark Abbot F
North Texas Eye Research Institute, University of North Texas Health Science Center, 3500 Camp Bowie Boulevard, Fort Worth, TX, 76109, USA.
Department of Pharmaceutical Sciences, University of North Texas Health Science Center, Fort Worth, TX, 76107, USA.
Mol Neurodegener. 2016 Apr 21;11:30. doi: 10.1186/s13024-016-0093-4.
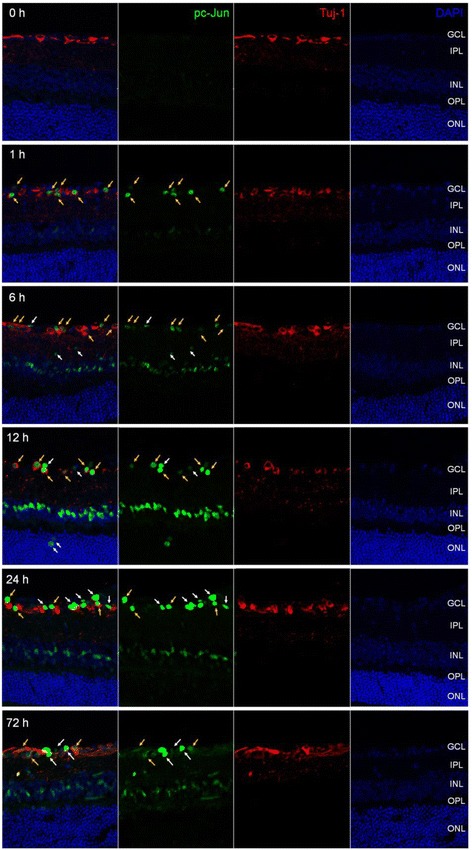
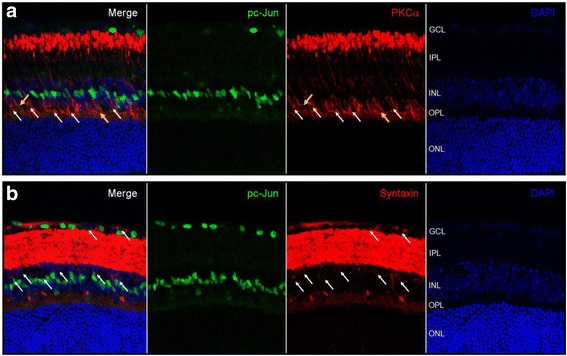
The c-Jun N-terminal kinase (JNK) signaling pathway plays an important role in neuronal pathophysiology. Using JNK inhibitors, we examined involvement of the JNK pathway in cultured rat retinal ganglion cell (RGC) death and in mouse retinal ischemia/reperfusion (I/R) injury of the visual axis. The in vitro effects of JNK inhibitors were evaluated in cultured adult rat retinal cells enriched in RGCs. Retinal I/R was induced in C57BL/6J mice through elevation of intraocular pressure to 120 mmHg for 60 min followed by reperfusion. SP600125 was administered intraperitoneally once daily for 28 days. Phosphorylation of JNK and c-Jun in the retina was examined by immunoblotting and immunohistochemistry. The thickness of retinal layers and cell numbers in the ganglion cell layer (GCL) were examined using H&E stained retinal cross sections and spectral domain optical coherence tomography (SD-OCT). Retinal function was measured by scotopic flash electroretinography (ERG). Volumetric measurement of the superior colliculus (SC) as well as VGLUT2 and PSD95 expression were studied.
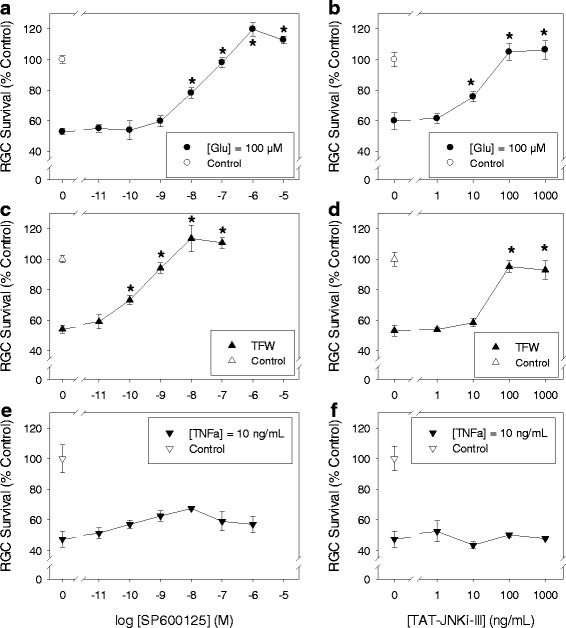
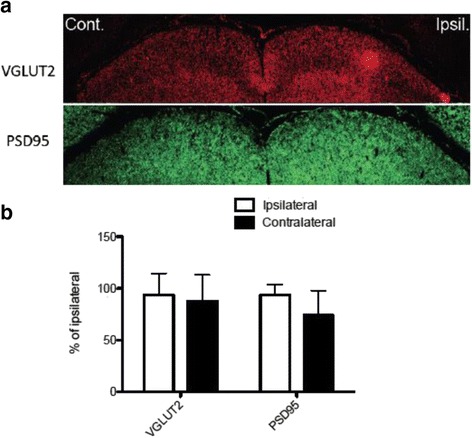
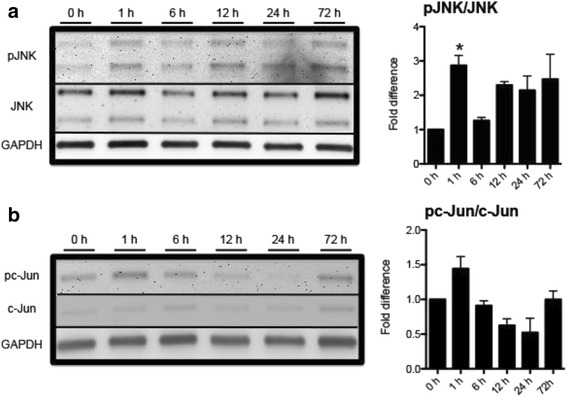
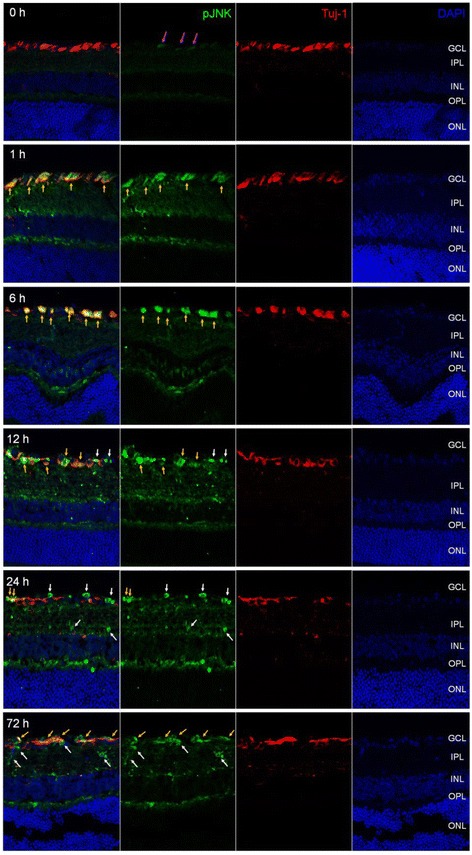
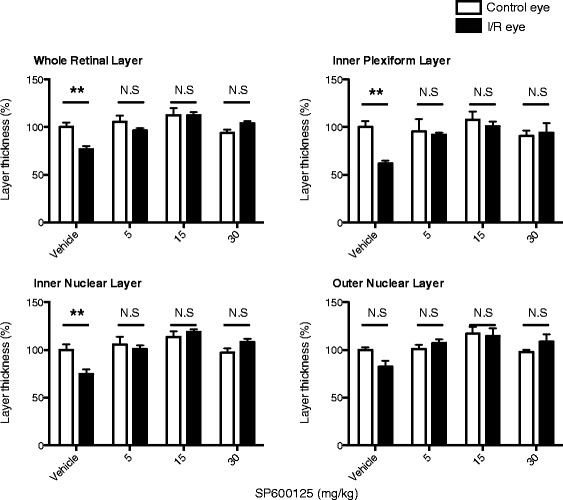
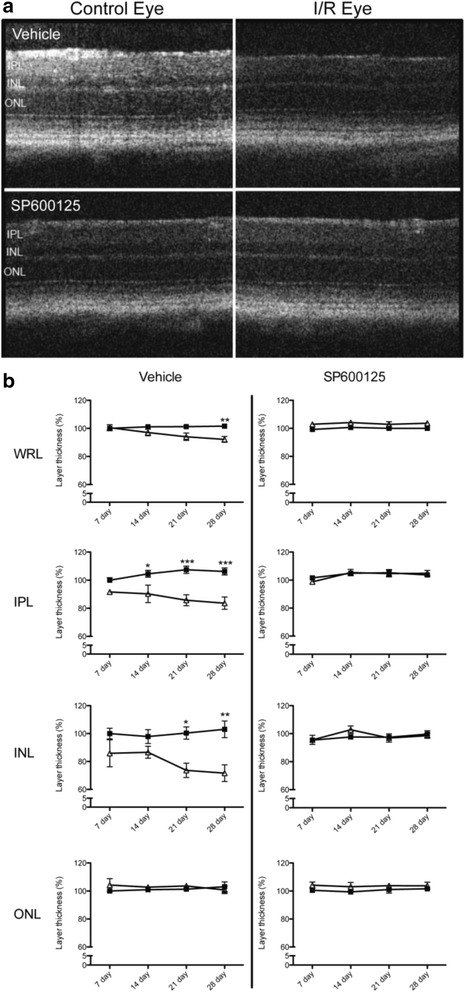
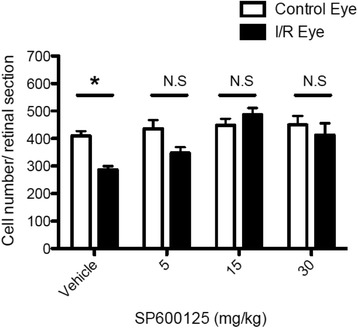
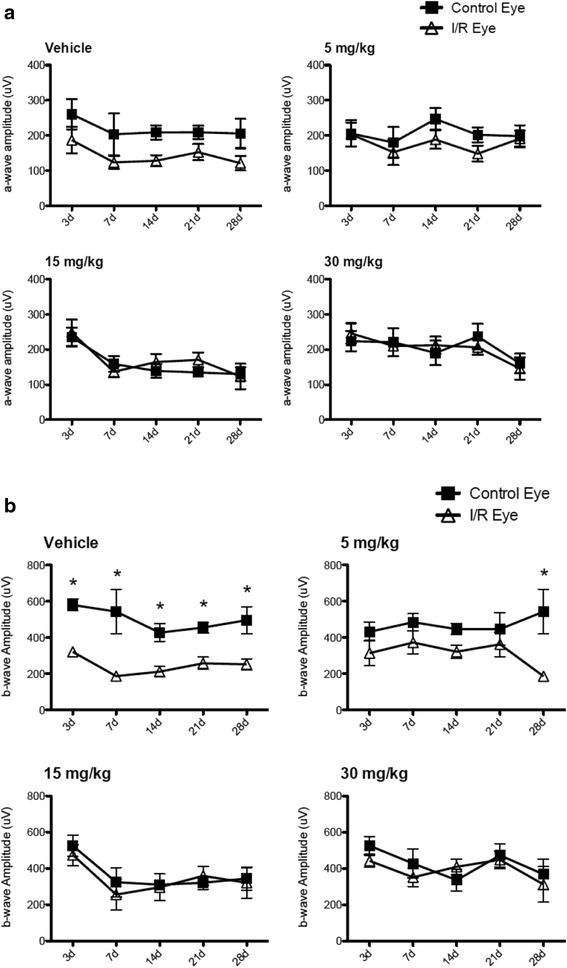
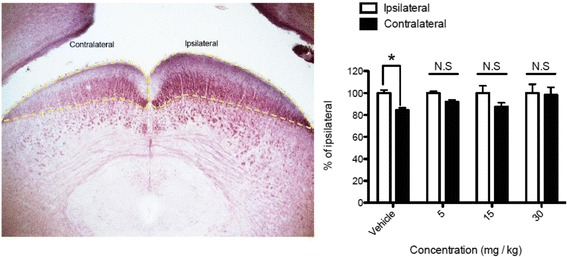
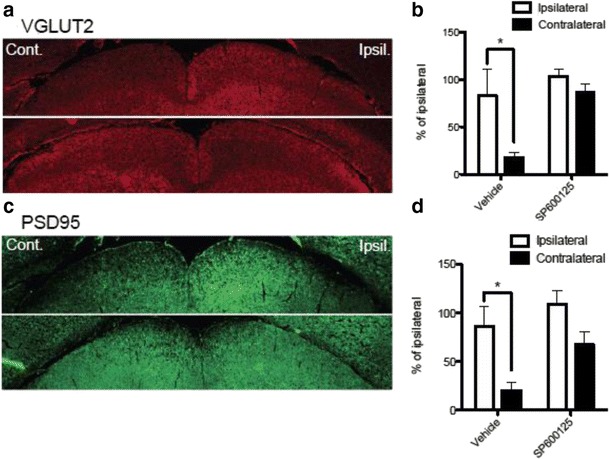
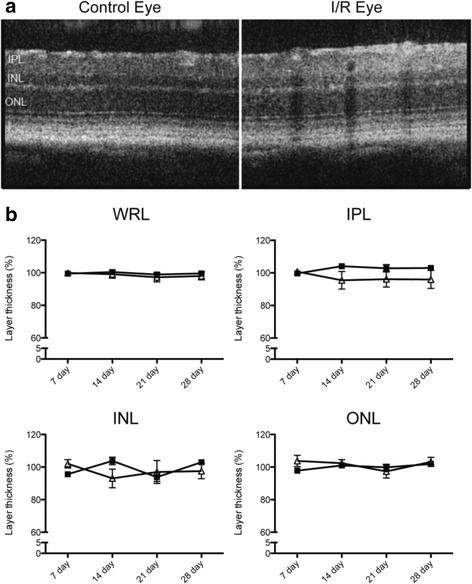
JNK inhibitors SP600125 and TAT-JNK-III, dose-dependently and significantly (p < 0.05) protected against glutamate excitotoxicity and trophic factor withdrawal induced RGC death in culture. In the I/R model, phosphorylation of JNK (pJNK) in the retina was significantly (p < 0.05) increased after injury. I/R injury significantly (p < 0.05) decreased the thickness of retinal layers, including the whole retina, inner plexiform layer, and inner nuclear layer and cell numbers in the GCL. Administration of SP600125 for 28 days protected against all these degenerative morphological changes (p < 0.05). In addition, SP600125 significantly (p < 0.05) protected against I/R-induced reduction in scotopic ERG b-wave amplitude at 3, 7, 14, 21 and 28 days after injury. SP600125 also protected against the I/R-induced losses in volume and levels of synaptic markers in the SC. Moreover, the protective effects of SP600125 in the retina and SC were also detected even with only 7 days (Days 1-7 after I/R) of SP600125 treatment.
Our results demonstrate the important role the JNK pathway plays in retinal degeneration in both in vitro and in vivo models and suggest that JNK inhibitors may be a useful therapeutic strategy for neuroprotection of RGCs in the retina.
c-Jun氨基末端激酶(JNK)信号通路在神经元病理生理学中起重要作用。我们使用JNK抑制剂,研究了JNK通路在培养的大鼠视网膜神经节细胞(RGC)死亡以及小鼠视轴视网膜缺血/再灌注(I/R)损伤中的作用。在富含RGC的成年大鼠视网膜细胞培养物中评估JNK抑制剂的体外作用。通过将眼压升高至120 mmHg持续60分钟,然后再灌注,在C57BL/6J小鼠中诱导视网膜I/R。每天腹腔注射SP600125一次,共28天。通过免疫印迹和免疫组织化学检测视网膜中JNK和c-Jun的磷酸化。使用苏木精和伊红(H&E)染色的视网膜横断面和光谱域光学相干断层扫描(SD-OCT)检查视网膜层的厚度和神经节细胞层(GCL)中的细胞数量。通过暗视闪光视网膜电图(ERG)测量视网膜功能。研究了上丘(SC)的体积测量以及VGLUT2和PSD95的表达。
JNK抑制剂SP600125和TAT-JNK-III在体外剂量依赖性且显著地(p<0.05)保护培养的RGC免受谷氨酸兴奋性毒性和营养因子剥夺诱导的死亡。在I/R模型中,损伤后视网膜中JNK(pJNK)的磷酸化显著(p<0.05)增加。I/R损伤显著(p<0.05)降低了包括整个视网膜、内网状层和内核层在内的视网膜层厚度以及GCL中的细胞数量。给予SP600125 28天可防止所有这些退行性形态学变化(p<0.05)。此外,SP600125在损伤后第3、7、14、21和28天显著(p<0.05)保护I/R诱导的暗视ERG b波振幅降低。SP600125还可防止I/R诱导的SC中突触标记物的体积和水平损失。此外,即使仅用SP600125治疗7天(I/R后的第1-7天),也能检测到SP600125对视网膜和SC的保护作用。
我们的结果证明了JNK通路在体外和体内模型的视网膜变性中所起的重要作用,并表明JNK抑制剂可能是视网膜中RGC神经保护的一种有用治疗策略。