Kromann Jimmy C, Christensen Anders S, Cui Qiang, Jensen Jan H
Department of Chemistry, University of Copenhagen , Copenhagen , Denmark.
Department of Chemistry, University of Wisconsin-Madison , Madison, WI , United States.
PeerJ. 2016 May 3;4:e1994. doi: 10.7717/peerj.1994. eCollection 2016.
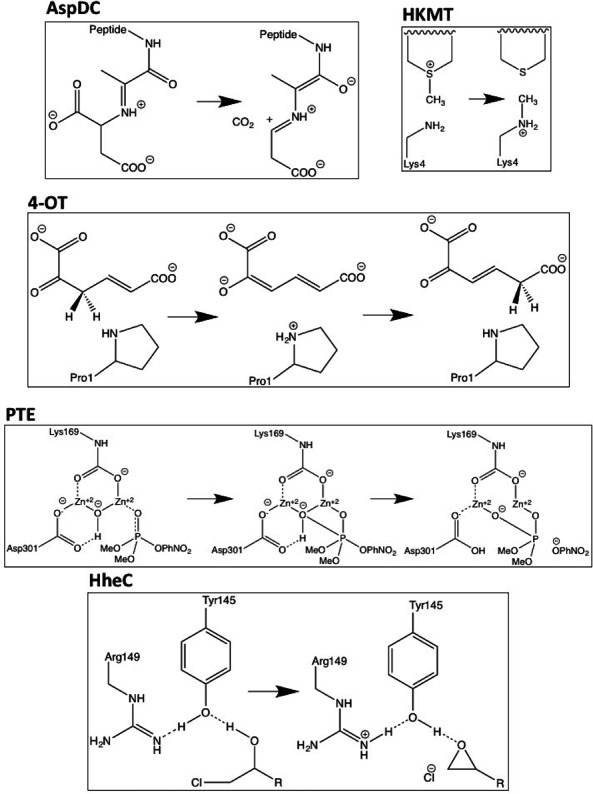
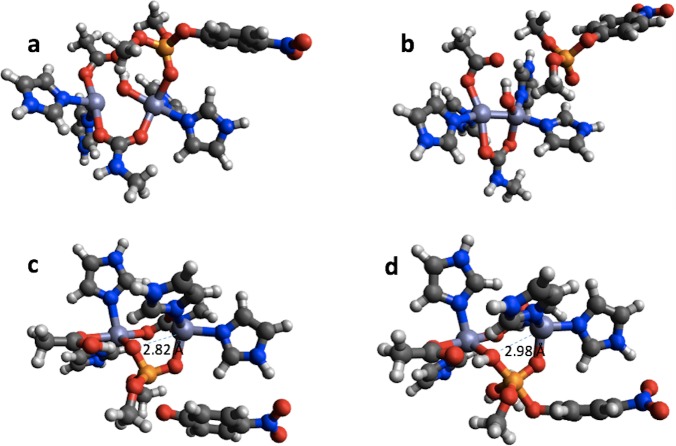
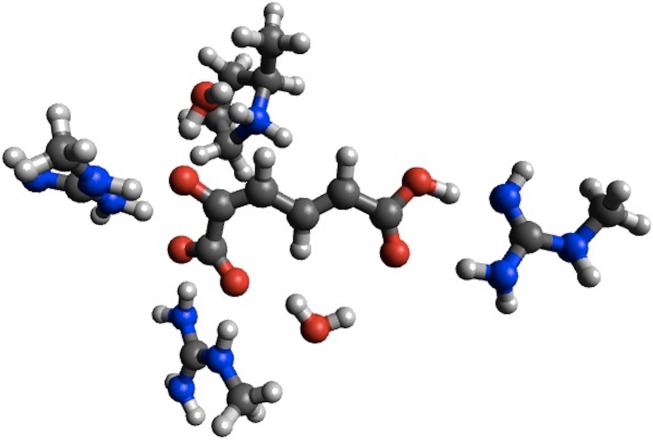
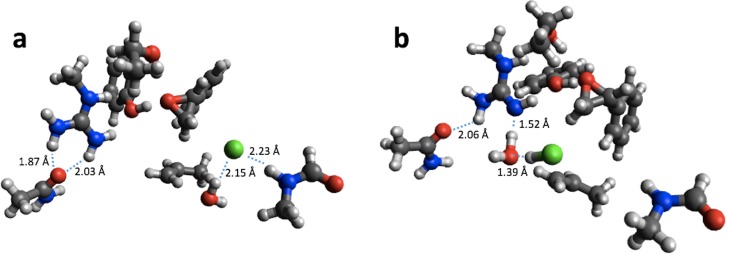
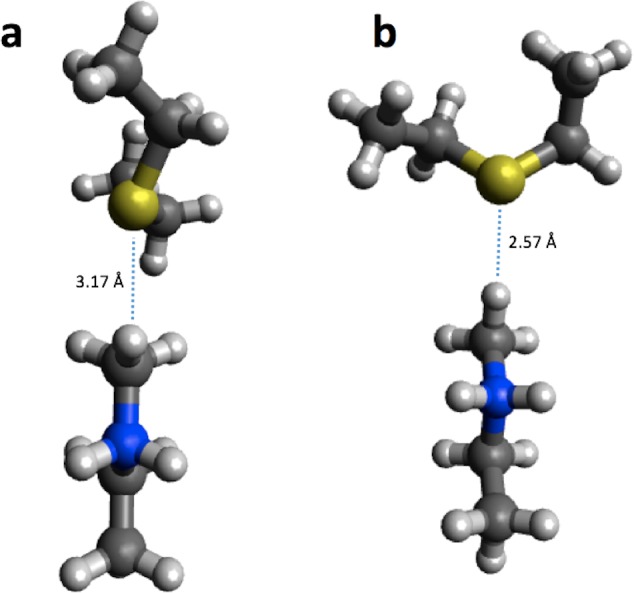
We have collected computed barrier heights and reaction energies (and associated model structures) for five enzymes from studies published by Himo and co-workers. Using this data, obtained at the B3LYP/6- 311+G(2d,2p)[LANL2DZ]//B3LYP/6-31G(d,p) level of theory, we then benchmark PM6, PM7, PM7-TS, and DFTB3 and discuss the influence of system size, bulk solvation, and geometry re-optimization on the error. The mean absolute differences (MADs) observed for these five enzyme model systems are similar to those observed for PM6 and PM7 for smaller systems (10-15 kcal/mol), while DFTB results in a MAD that is significantly lower (6 kcal/mol). The MADs for PMx and DFTB3 are each dominated by large errors for a single system and if the system is disregarded the MADs fall to 4-5 kcal/mol. Overall, results for the condensed phase are neither more or less accurate relative to B3LYP than those in the gas phase. With the exception of PM7-TS, the MAD for small and large structural models are very similar, with a maximum deviation of 3 kcal/mol for PM6. Geometry optimization with PM6 shows that for one system this method predicts a different mechanism compared to B3LYP/6-31G(d,p). For the remaining systems, geometry optimization of the large structural model increases the MAD relative to single points, by 2.5 and 1.8 kcal/mol for barriers and reaction energies. For the small structural model, the corresponding MADs decrease by 0.4 and 1.2 kcal/mol, respectively. However, despite these small changes, significant changes in the structures are observed for some systems, such as proton transfer and hydrogen bonding rearrangements. The paper represents the first step in the process of creating a benchmark set of barriers computed for systems that are relatively large and representative of enzymatic reactions, a considerable challenge for any one research group but possible through a concerted effort by the community. We end by outlining steps needed to expand and improve the data set and how other researchers can contribute to the process.
我们从希莫及其同事发表的研究中收集了五种酶的计算势垒高度和反应能量(以及相关的模型结构)。利用在B3LYP/6 - 311+G(2d,2p)[LANL2DZ]//B3LYP/6 - 31G(d,p)理论水平下获得的数据,我们随后对PM6、PM7、PM7 - TS和DFTB3进行了基准测试,并讨论了体系大小、整体溶剂化和几何结构重新优化对误差的影响。在这五个酶模型体系中观察到的平均绝对差值(MAD)与在较小体系(10 - 15千卡/摩尔)中PM6和PM7观察到的相似,而DFTB的MAD显著更低(6千卡/摩尔)。PMx和DFTB3的MAD均由单个体系的大误差主导,如果忽略该体系,MAD降至4 - 5千卡/摩尔。总体而言,凝聚相的结果相对于B3LYP而言,在气相中既没有更准确也没有更不准确。除了PM7 - TS,小结构模型和大结构模型的MAD非常相似,PM6的最大偏差为3千卡/摩尔。用PM6进行几何结构优化表明,对于一个体系,该方法预测的机理与B3LYP/6 - 31G(d,p)不同。对于其余体系,大结构模型的几何结构优化使相对于单点计算的MAD在势垒和反应能量方面分别增加了2.5和1.8千卡/摩尔。对于小结构模型,相应的MAD分别降低了0.4和1.2千卡/摩尔。然而,尽管有这些小的变化,但在一些体系中仍观察到结构的显著变化,如质子转移和氢键重排。本文代表了为相对较大且代表酶促反应的体系创建计算势垒基准集过程的第一步,这对任何一个研究小组来说都是一项相当大的挑战,但通过社区的共同努力是可能的。我们最后概述了扩展和改进数据集所需的步骤以及其他研究人员如何为该过程做出贡献。