Goodwin Sara, McPherson John D, McCombie W Richard
Cold Spring Harbor Laboratory, Cold Spring Harbor, New York 11724, USA.
Department of Biochemistry and Molecular Medicine; and the Comprehensive Cancer Center, University of California, Davis, California 95817, USA.
Nat Rev Genet. 2016 May 17;17(6):333-51. doi: 10.1038/nrg.2016.49.
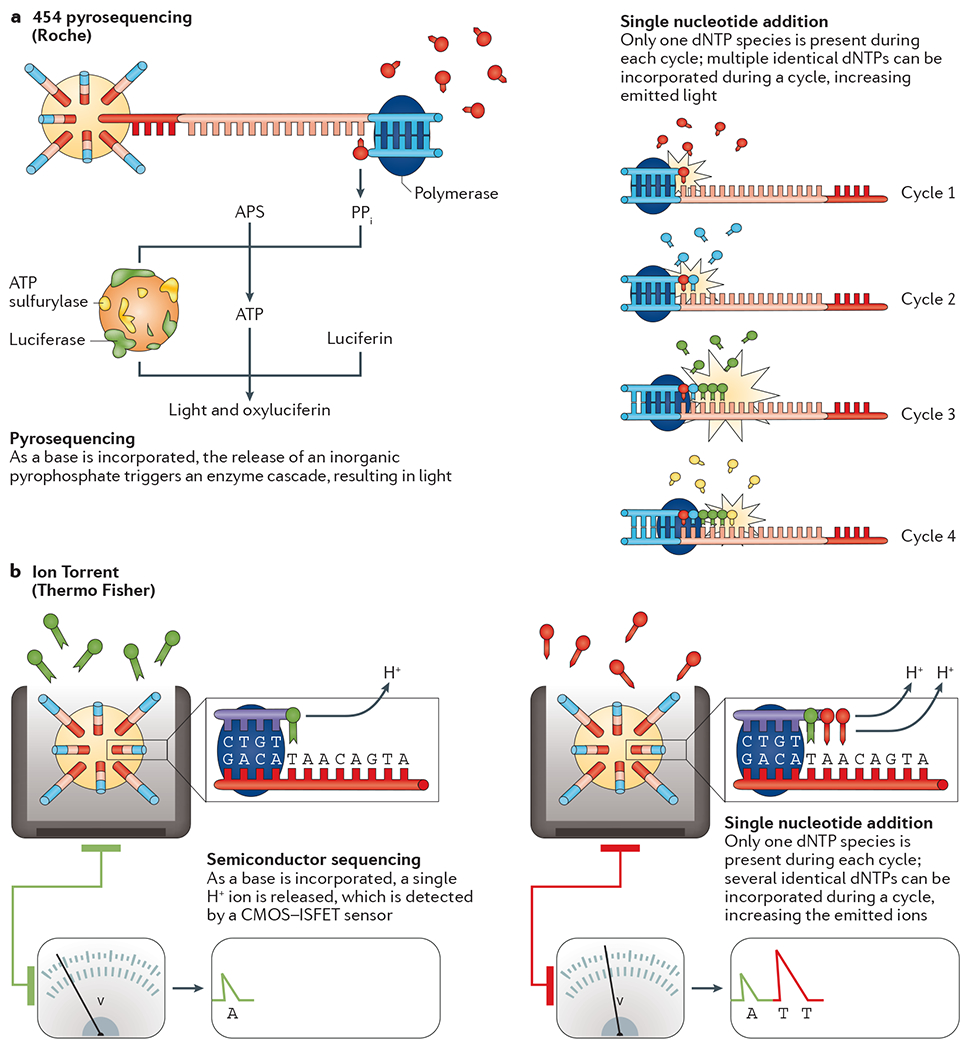
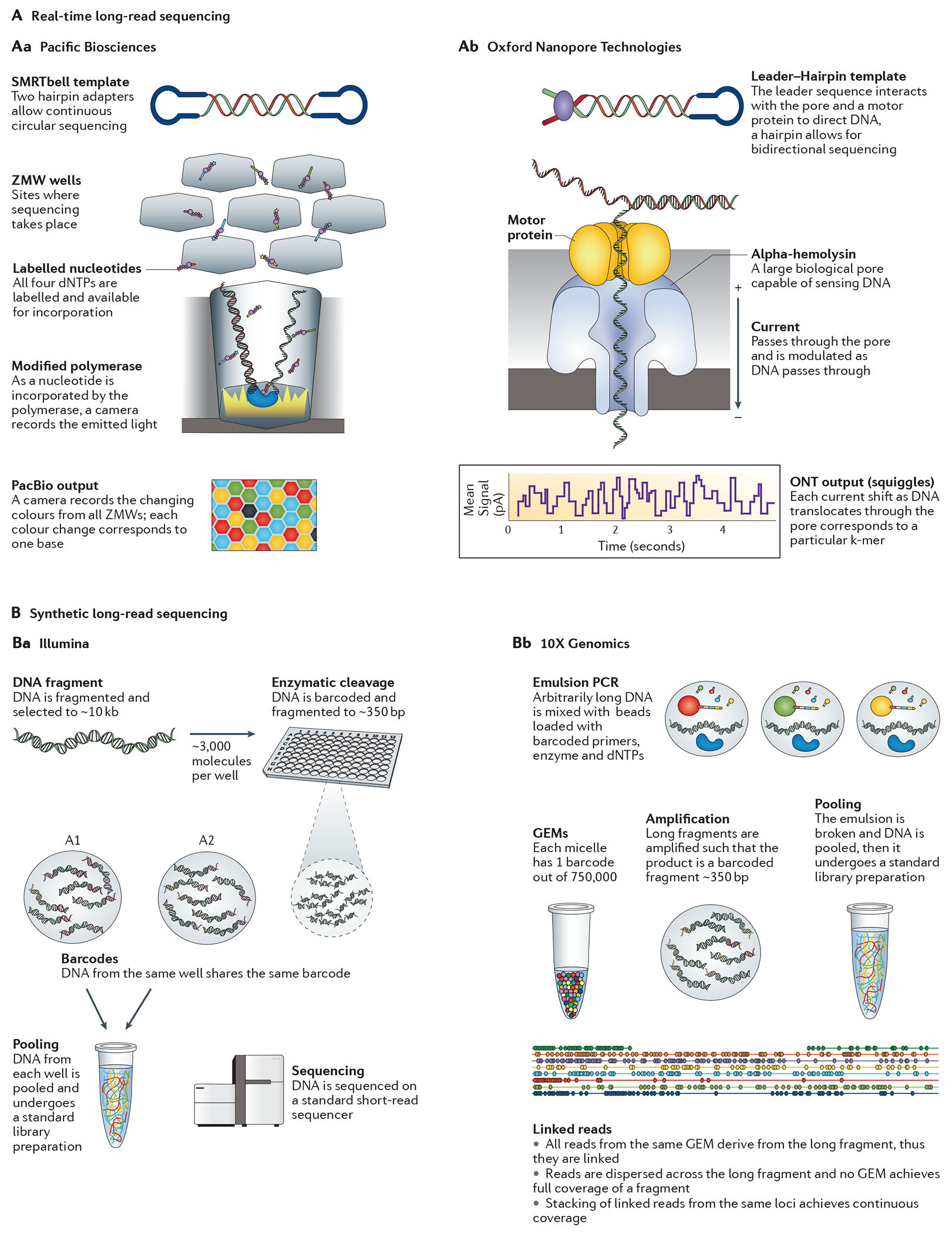
Since the completion of the human genome project in 2003, extraordinary progress has been made in genome sequencing technologies, which has led to a decreased cost per megabase and an increase in the number and diversity of sequenced genomes. An astonishing complexity of genome architecture has been revealed, bringing these sequencing technologies to even greater advancements. Some approaches maximize the number of bases sequenced in the least amount of time, generating a wealth of data that can be used to understand increasingly complex phenotypes. Alternatively, other approaches now aim to sequence longer contiguous pieces of DNA, which are essential for resolving structurally complex regions. These and other strategies are providing researchers and clinicians a variety of tools to probe genomes in greater depth, leading to an enhanced understanding of how genome sequence variants underlie phenotype and disease.
自2003年人类基因组计划完成以来,基因组测序技术取得了非凡进展,这使得每兆碱基的成本降低,测序基因组的数量和多样性增加。基因组结构惊人的复杂性已被揭示,推动这些测序技术取得更大进步。一些方法在最短时间内使测序碱基数量最大化,生成大量可用于理解日益复杂表型的数据。或者,其他方法现在旨在对更长的连续DNA片段进行测序,这对于解析结构复杂区域至关重要。这些及其他策略为研究人员和临床医生提供了各种工具,以便更深入地探测基因组,从而增强对基因组序列变异如何构成表型和疾病基础的理解。