Kim Nam Hee, Livi Carolina B, Yew P Renee, Boyer Thomas G
Department of Molecular Medicine and Institute of Biotechnology, The University of Texas Health Science Center at San Antonio, San Antonio, TX, 78229-3900, USA.
Agilent Technologies, Portland, OR, 97224-7154, USA.
BMC Dev Biol. 2016 May 17;16(1):17. doi: 10.1186/s12861-016-0114-0.
The RNA polymerase II transcriptional Mediator subunit Med12 is broadly implicated in vertebrate brain development, and genetic variation in human MED12 is associated with X-linked intellectual disability and neuropsychiatric disorders. Although prior studies have begun to elaborate the functional contribution of Med12 within key neurodevelopmental pathways, a more complete description of Med12 function in the developing nervous system, including the specific biological networks and cellular processes under its regulatory influence, remains to be established. Herein, we sought to clarify the global contribution of Med12 to neural stem cell (NSC) biology through unbiased transcriptome profiling of mouse embryonic stem (ES) cell-derived NSCs following RNAi-mediated Med12 depletion.
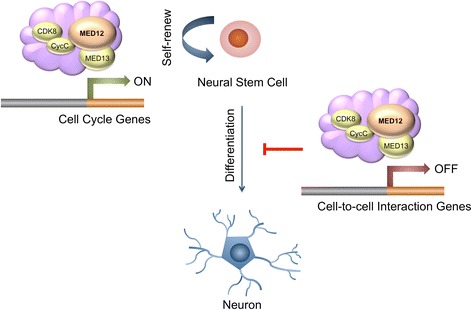
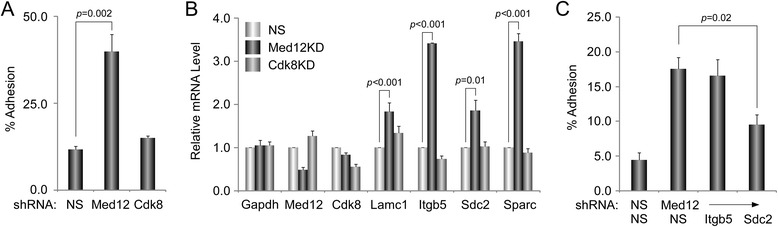
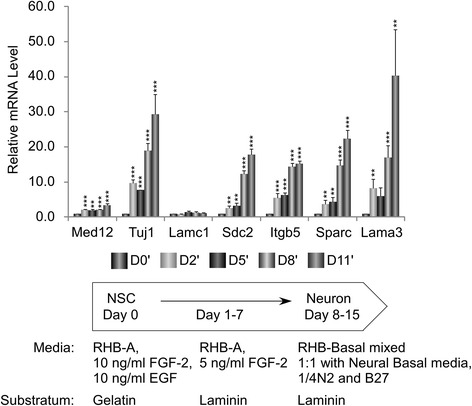
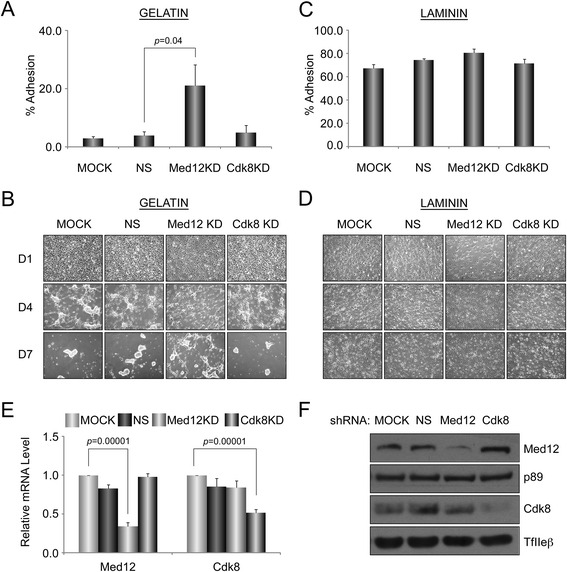
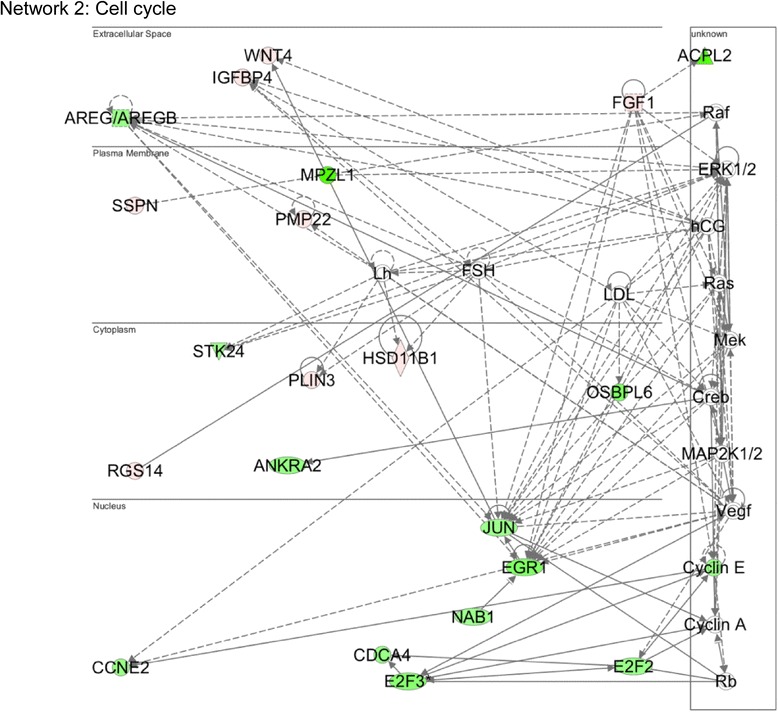
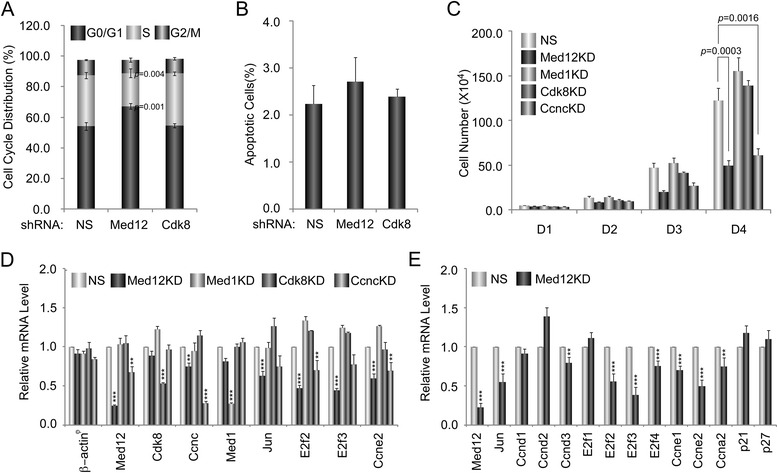
A total of 240 genes (177 up, 73 down) were differentially expressed in Med12-knockdown versus control mouse NS-5 (mNS-5) NSCs. Gene set enrichment analysis revealed Med12 to be prominently linked with "cell-to-cell interaction" and "cell cycle" networks, and subsequent functional studies confirmed these associations. Targeted depletion of Med12 led to enhanced NSC adhesion and upregulation of cell adhesion genes, including Syndecan 2 (Sdc2). Concomitant depletion of both Sdc2 and Med12 reversed enhanced cell adhesion triggered by Med12 knockdown alone, confirming that Med12 negatively regulates NSC cell adhesion by suppressing the expression of cell adhesion molecules. Med12-mediated suppression of NSC adhesion is a dynamically regulated process in vitro, enforced in self-renewing NSCs and alleviated during the course of neuronal differentiation. Accordingly, Med12 depletion enhanced adhesion and prolonged survival of mNS-5 NSCs induced to differentiate on gelatin, effects that were bypassed completely by growth on laminin. On the other hand, Med12 depletion in mNS-5 NSCs led to reduced expression of G1/S phase cell cycle regulators and a concordant G1/S phase cell cycle block without evidence of apoptosis, resulting in a severe proliferation defect.
Med12 contributes to the maintenance of NSC identity through a functionally bipartite role in suppression and activation of gene expression programs dedicated to cell adhesion and G1/S phase cell cycle progression, respectively. Med12 may thus contribute to the regulatory apparatus that controls the balance between NSC self-renewal and differentiation, with important implications for MED12-linked neurodevelopmental disorders.
RNA聚合酶II转录中介体亚基Med12广泛参与脊椎动物脑发育,人类MED12基因变异与X连锁智力残疾和神经精神疾病相关。尽管先前的研究已开始阐述Med12在关键神经发育途径中的功能作用,但对于Med12在发育中的神经系统中的功能,包括其调控影响下的特定生物网络和细胞过程,仍有待更全面的描述。在此,我们试图通过对RNAi介导的Med12缺失后小鼠胚胎干细胞(ES)来源的神经干细胞(NSC)进行无偏转录组分析,来阐明Med12对神经干细胞生物学的整体贡献。
在Med12敲低的小鼠NS-5(mNS-5)神经干细胞与对照相比,共有240个基因(177个上调,73个下调)差异表达。基因集富集分析显示Med12与“细胞间相互作用”和“细胞周期”网络显著相关,随后的功能研究证实了这些关联。靶向缺失Med12导致神经干细胞黏附增强以及细胞黏附基因上调,包括Syndecan 2(Sdc2)。同时缺失Sdc2和Med12可逆转仅由Med12敲低引发的细胞黏附增强,证实Med12通过抑制细胞黏附分子的表达来负向调节神经干细胞的细胞黏附。Med12介导的神经干细胞黏附抑制在体外是一个动态调节过程,在自我更新的神经干细胞中起作用,而在神经元分化过程中减弱。因此,Med12缺失增强了在明胶上诱导分化的mNS-5神经干细胞的黏附并延长了其存活时间,而在层粘连蛋白上生长则完全消除了这些影响。另一方面,mNS-5神经干细胞中Med12缺失导致G1/S期细胞周期调节因子表达降低以及一致的G1/S期细胞周期阻滞,且无凋亡证据,从而导致严重的增殖缺陷。
Med12通过在分别抑制和激活与细胞黏附及G1/S期细胞周期进程相关的基因表达程序中发挥双重功能,来维持神经干细胞的特性。因此,Med12可能参与了控制神经干细胞自我更新与分化平衡的调节机制,这对与MED12相关的神经发育障碍具有重要意义。