Beck Bodo B, van Spronsen FrancJan, Diepstra Arjan, Berger Rolf M F, Kömhoff Martin
Institute of Human Genetics, University of Cologne, Cologne, Germany.
Division of Metabolic Diseases, Beatrix Children's Hospital, University Medical Centre Groningen, University of Groningen, Groningen, The Netherlands.
Pediatr Nephrol. 2017 May;32(5):733-741. doi: 10.1007/s00467-016-3399-0. Epub 2016 Jun 11.
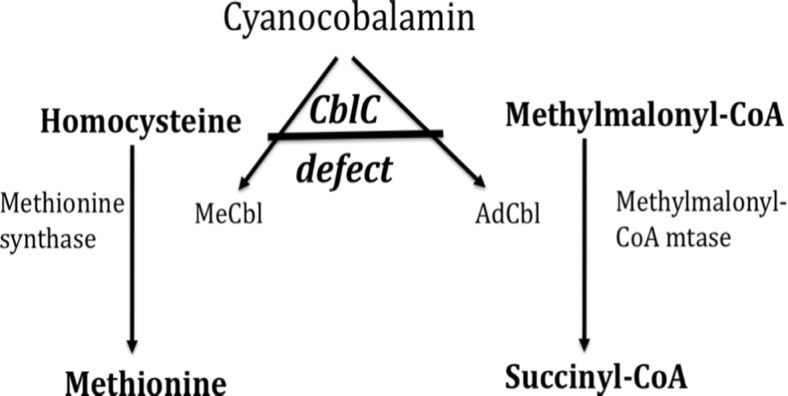
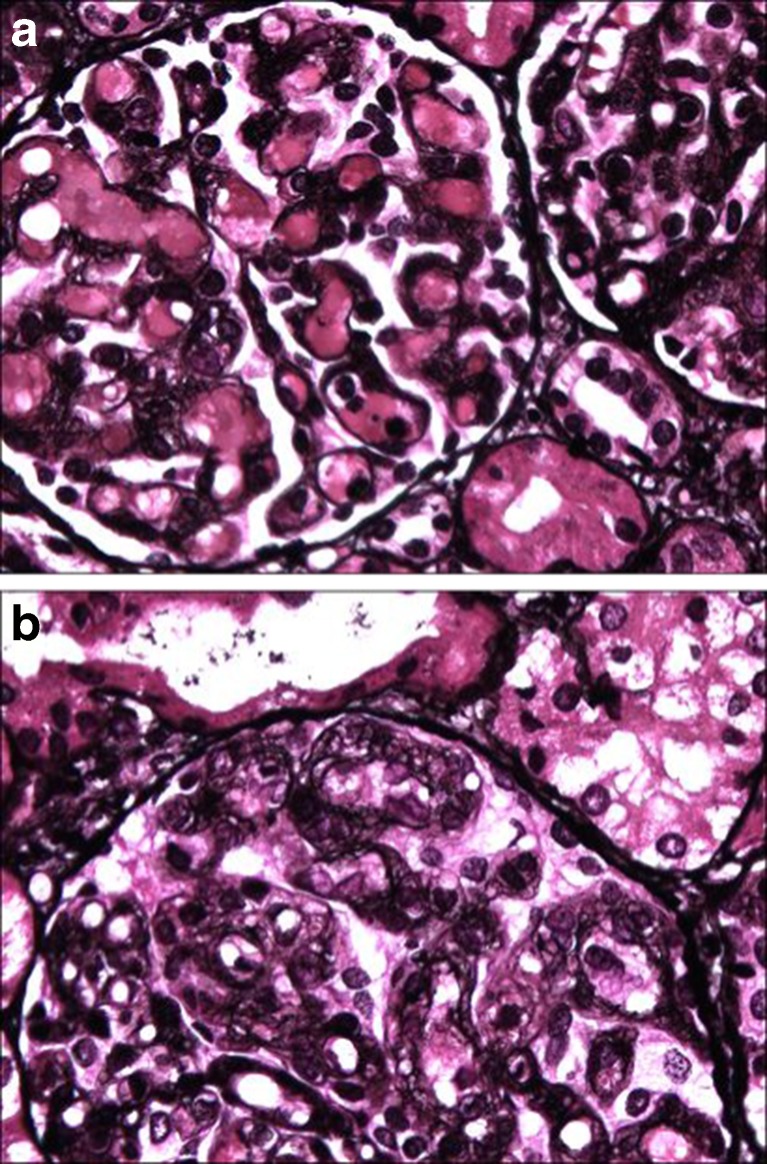
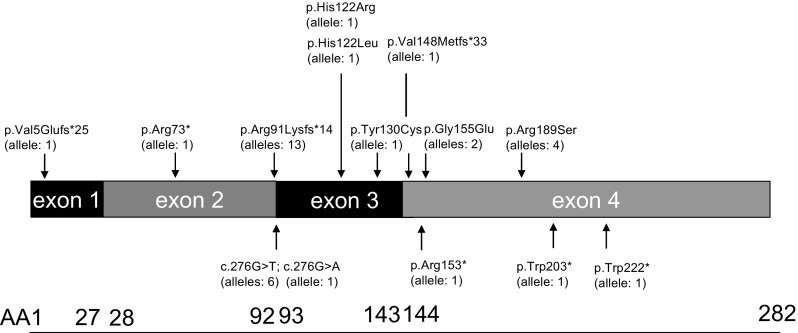
Methylmalonic aciduria and homocystinuria, cobalamin C (cblC) type, is the most common genetic type of functional cobalamin (vitamin B) deficiency. This metabolic disease is characterized by marked heterogeneity of neurocognitive disease (microcephaly, seizures, developmental delay, ataxia, hypotonia) and variable extracentral nervous system involvement (failure to thrive, cardiovascular, renal, ocular) manifesting predominantly early in life, sometimes during gestation. To enhance awareness and understanding of renal disease associated with cblC defect, we studied biochemical, genetic, clinical, and histopathological data from 36 patients. Consistent clinical chemistry features of renal disease were intravascular hemolysis, hematuria, and proteinuria in all patients, with nephrotic-range proteinuria observed in three. Renal function ranged from normal to renal failure, with eight patients requiring (intermittent) dialysis. Two thirds were diagnosed with atypical (diarrhea-negative) hemolytic uremic syndrome (HUS). Renal histopathology analyses of biopsy samples from 16 patients revealed glomerular lesions typical of thrombotic microangiopathy (TMA). Treatment with hydroxycobalamin improved renal function in the majority, including three in whom dialysis could be withdrawn. Neurological sequelae were observed in 44 % and cardiopulmonary involvement in 39 % of patients, with half of the latter group demonstrating pulmonary hypertension. Mortality reached 100 % in untreated patients and 79 and 56 % in those with cardiopulmonary or neurological involvement, respectively. In all patients presenting with unclear intravascular hemolysis, hematuria, and proteinuria, cblC defect should be ruled out by determination of blood/plasma homocysteine levels and/or genetic testing, irrespective of actual renal function and neurological status, to ensure timely diagnosis and treatment.
甲基丙二酸尿症和同型胱氨酸尿症,钴胺素C(cblC)型,是功能性钴胺素(维生素B)缺乏最常见的遗传类型。这种代谢疾病的特征是神经认知疾病(小头畸形、癫痫发作、发育迟缓、共济失调、肌张力减退)具有明显的异质性,以及可变的中枢神经系统外受累情况(生长发育迟缓、心血管、肾脏、眼部),主要在生命早期出现,有时在妊娠期出现。为了提高对与cblC缺陷相关的肾脏疾病的认识和理解,我们研究了36例患者的生化、遗传、临床和组织病理学数据。肾脏疾病一致的临床化学特征是所有患者均有血管内溶血、血尿和蛋白尿,其中3例出现肾病范围的蛋白尿。肾功能从正常到肾衰竭不等,8例患者需要(间歇性)透析。三分之二的患者被诊断为非典型(腹泻阴性)溶血性尿毒症综合征(HUS)。对16例患者的活检样本进行肾脏组织病理学分析,发现了血栓性微血管病(TMA)典型的肾小球病变。大多数患者使用羟钴胺治疗后肾功能得到改善,其中3例患者可以停止透析。44%的患者出现神经后遗症,39%的患者出现心肺受累,后一组中有一半表现为肺动脉高压。未经治疗的患者死亡率达到100%,有心肺或神经受累的患者死亡率分别为79%和56%。对于所有出现不明原因的血管内溶血、血尿和蛋白尿的患者,无论实际肾功能和神经状态如何,都应通过测定血液/血浆同型半胱氨酸水平和/或基因检测排除cblC缺陷,以确保及时诊断和治疗。