Liu Xiaoyu, Xiao Huijie, Yao Yong, Wang Suxia, Zhang Hongwen, Zhong Xuhui, Yang Yanling, Ding Jie, Wang Fang
Department of Pediatrics, Peking University First Hospital, Beijing, China.
Laboratory of Electron Microscopy, Pathological Center, Peking University First Hospital, Beijing, China.
Front Pediatr. 2023 Jan 10;10:1057594. doi: 10.3389/fped.2022.1057594. eCollection 2022.
CblC deficiency, the most common cobalamin metabolic abnormality, is caused by pathogenic variants in the gene. The renal complications of this disease have been described only in a small number of cases. This study aimed to better delineate renal phenotype and genetic characteristics in Chinese children with cblC defect.
Children with cblC deficiency who manifested as kidney damage were enrolled. Clinical, renal pathological, and genetic data were reviewed in detail.
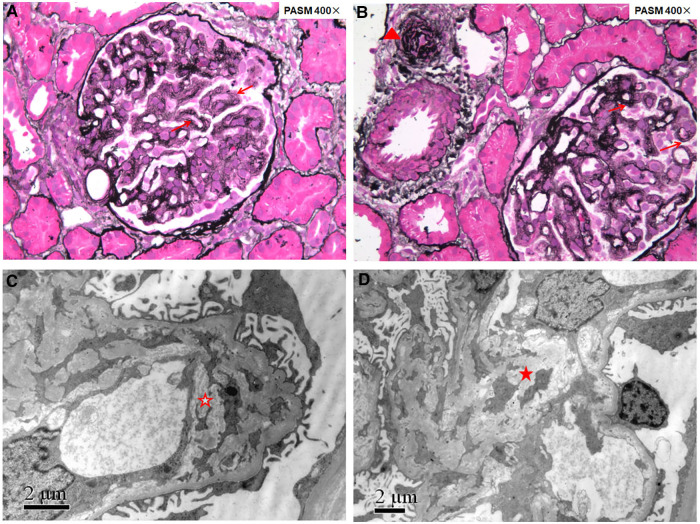
Seven cases were enrolled. Ages at disease onset ranged from 9 months to 5 years. All patients presented with hematuria and proteinuria, and 2/7 cases presented with nephrotic syndrome. Renal dysfunction was observed in 4/7 cases. Renal biopsy was performed in 5/7 cases, and all of them had renal thrombotic microangiopathy. Macrocytic anemia was detected in all seven patients. Six out of seven cases had hypertension, and 2/7 cases presented with pulmonary hypertension. Two of them had a mild intellectual disability, and one suffered from epilepsy. Increased urine methylmalonic acid and plasma homocysteine were detected in seven cases, while two patients had normal levels of urine methylmalonic acid at the initial evaluation. After diagnosis, all seven cases were treated with hydroxocobalamin IM. Six cases were followed-up for 3-8 years. After treatments, anemia was the first to be recovered, followed by proteinuria. Renal function recovered after 1 year in two cases, whereas patient 2 progressed to stage 2 chronic kidney disease 13 years after onset. While a case presented with end-stage kidney disease because of late diagnosis, one case died 3 months after disease onset due to giving up treatment. Three pathogenic variants c.80A > G (8/14), c.609G > A (4/14), and c.658_660delAAG (2/14) were detected in all seven children.
variant c.80A > G may be associated with prominent renal complications in Chinese cblC patients. Macrocytic anemia and hyperhomocysteinemia are useful clues for patients with hematuria and proteinuria caused by cblC defect. The most frequent renal pathological manifestation is thrombotic microangiopathy. Early diagnosis and treatment resulted in improving renal and hematological signs.
CblC缺陷是最常见的钴胺素代谢异常,由该基因的致病变异引起。这种疾病的肾脏并发症仅在少数病例中有所描述。本研究旨在更好地描述中国cblC缺陷儿童的肾脏表型和遗传特征。
纳入表现为肾脏损害的cblC缺陷儿童。详细回顾临床、肾脏病理和遗传数据。
共纳入7例。发病年龄为9个月至5岁。所有患者均有血尿和蛋白尿,2/7例表现为肾病综合征。4/7例观察到肾功能不全。5/7例行肾脏活检,均有肾脏血栓性微血管病。所有7例患者均检测到大细胞贫血。7例中有6例高血压,2/7例有肺动脉高压。其中2例有轻度智力障碍,1例患有癫痫。7例患者尿甲基丙二酸和血浆同型半胱氨酸升高,2例患者初诊时尿甲基丙二酸水平正常。确诊后,所有7例均接受羟钴胺素肌肉注射治疗。6例随访3至8年。治疗后,贫血最先恢复,其次是蛋白尿。2例患者肾功能在1年后恢复,而患者2在发病13年后进展为2期慢性肾脏病。1例因诊断延迟出现终末期肾病,1例因放弃治疗在发病3个月后死亡。在所有7名儿童中检测到3种致病变异c.80A>G(8/14)、c.609G>A(4/14)和c.658_660delAAG(2/14)。
变异型c.80A>G可能与中国cblC患者显著的肾脏并发症有关。大细胞贫血和高同型半胱氨酸血症是cblC缺陷导致血尿和蛋白尿患者的有用线索。最常见的肾脏病理表现是血栓性微血管病。早期诊断和治疗有助于改善肾脏和血液学症状。