Renal Division, Department of Medicine, Peking University First Hospital, No. 8 Xishiku St., Xicheng District, Beijing, 100034, China.
Institute of Nephrology, Peking University, Beijing, China.
BMC Nephrol. 2024 Oct 10;25(1):340. doi: 10.1186/s12882-024-03774-w.
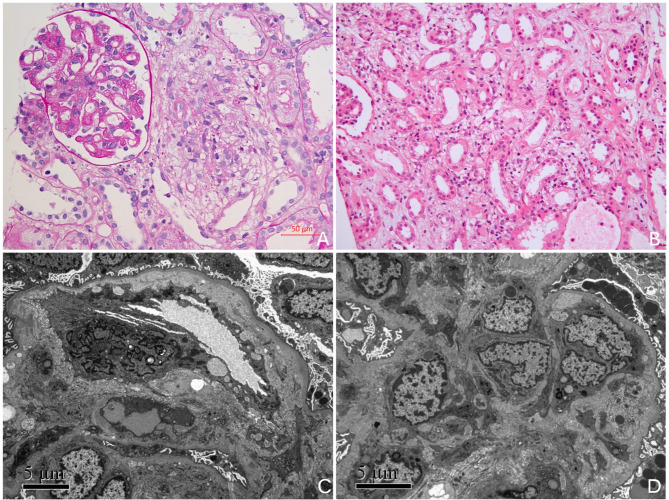
Mutation of MMACHC gene causes cobalamin C disease (cblC), an inherited metabolic disorder, which presents as combined methylmalonic aciduria (MMA-uria) and hyperhomocysteinaemia in clinical. Renal complications may also be present in patients with this inborn deficiency. The most common histological change is thrombotic microangiopathy (TMA). However, to our acknowledge, renal tubular injury in the late-onset presentation of cblC is rarely been reported. This study provides a detailed description of the characteristics of kidney disease in cblC deficiency, aiming to improve the early recognition of this treatable disease for clinical nephrologists.
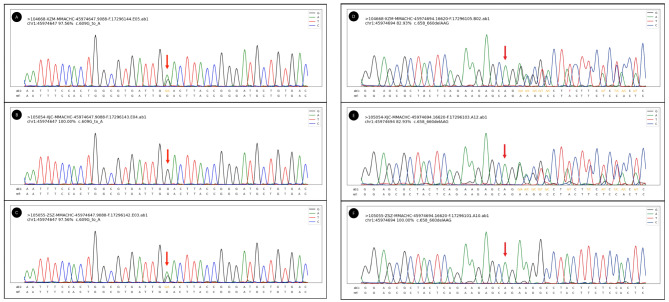
Here we described three teenage patients who presented with hematuria, proteinuria, and hypertension in clinical presentation. They were diagnosed with renal involvement due to cblC deficiency after laboratory tests revealing elevated serum and urine homocysteine, renal biopsy showing TMA and tubular injury, along with genetic testing showing heterogeneous compound mutations in MMACHC. Hydroxocobalamin, betaine, and L-carnitine were administered to these patients. All of them got improved, with decreased homocysteine, controlled blood pressure, and kidney outcomes recovered.
The clinical diagnosis of cblC disease associated with kidney injury should be considered in patients with unclear TMA accompanied by a high concentration of serum homocysteine, even in teenagers or adults. Early diagnosis and timely intervention are vital to improving the prognosis of cobalamin C disease.
Not applicable.
MMACHC 基因突变导致钴胺素 C 病(cblC),这是一种遗传性代谢疾病,在临床上表现为甲基丙二酸血症(MMA-uria)和高同型半胱氨酸血症合并存在。此类先天性缺陷的患者也可能出现肾脏并发症。最常见的组织学改变是血栓性微血管病(TMA)。然而,据我们所知,cblC 迟发性表现中的肾小管损伤很少有报道。本研究详细描述了 cblC 缺乏症患者肾脏疾病的特征,旨在提高临床肾病医生对这种可治疗疾病的早期认识。
这里我们描述了 3 名青少年患者,他们在临床上表现为血尿、蛋白尿和高血压。在实验室检查发现血清和尿液同型半胱氨酸升高、肾活检显示 TMA 和肾小管损伤,以及基因检测显示 MMACHC 存在异质性复合突变后,这些患者被诊断为 cblC 缺乏引起的肾损害。这些患者接受了羟钴胺、甜菜碱和左旋肉碱治疗。他们的病情都得到了改善,同型半胱氨酸降低,血压得到控制,肾脏结果得到恢复。
对于伴有高血清同型半胱氨酸的不明原因 TMA 患者,即使是青少年或成年人,也应考虑与肾脏损伤相关的 cblC 疾病的临床诊断。早期诊断和及时干预对改善钴胺素 C 病的预后至关重要。
不适用。