Ponomarenko Elena A, Poverennaya Ekaterina V, Ilgisonis Ekaterina V, Pyatnitskiy Mikhail A, Kopylov Arthur T, Zgoda Victor G, Lisitsa Andrey V, Archakov Alexander I
Institute of Biomedical Chemistry, Moscow 119121, Russia.
Int J Anal Chem. 2016;2016:7436849. doi: 10.1155/2016/7436849. Epub 2016 May 19.
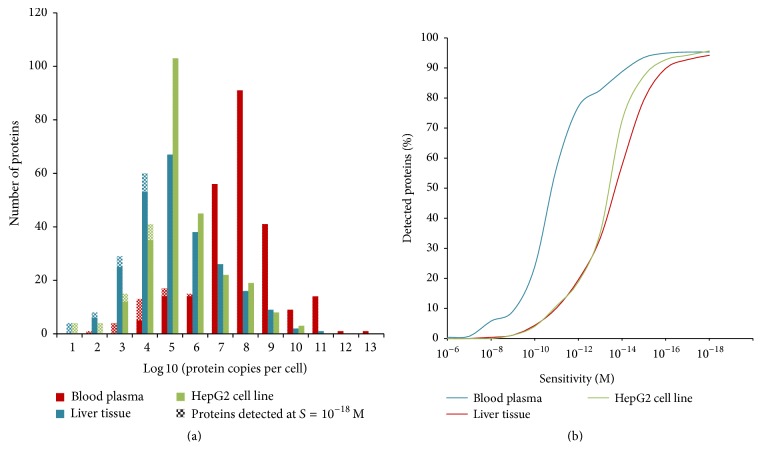
This work discusses bioinformatics and experimental approaches to explore the human proteome, a constellation of proteins expressed in different tissues and organs. As the human proteome is not a static entity, it seems necessary to estimate the number of different protein species (proteoforms) and measure the number of copies of the same protein in a specific tissue. Here, meta-analysis of neXtProt knowledge base is proposed for theoretical prediction of the number of different proteoforms that arise from alternative splicing (AS), single amino acid polymorphisms (SAPs), and posttranslational modifications (PTMs). Three possible cases are considered: (1) PTMs and SAPs appear exclusively in the canonical sequences of proteins, but not in splice variants; (2) PTMs and SAPs can occur in both proteins encoded by canonical sequences and in splice variants; (3) all modification types (AS, SAP, and PTM) occur as independent events. Experimental validation of proteoforms is limited by the analytical sensitivity of proteomic technology. A bell-shaped distribution histogram was generated for proteins encoded by a single chromosome, with the estimation of copy numbers in plasma, liver, and HepG2 cell line. The proposed metabioinformatics approaches can be used for estimation of the number of different proteoforms for any group of protein-coding genes.
这项工作讨论了探索人类蛋白质组的生物信息学和实验方法,人类蛋白质组是在不同组织和器官中表达的一组蛋白质。由于人类蛋白质组不是一个静态实体,因此似乎有必要估计不同蛋白质种类(蛋白质异构体)的数量,并测量特定组织中同一蛋白质的拷贝数。在此,提出对neXtProt知识库进行荟萃分析,以从理论上预测由可变剪接(AS)、单氨基酸多态性(SAP)和翻译后修饰(PTM)产生的不同蛋白质异构体的数量。考虑了三种可能的情况:(1)PTM和SAP仅出现在蛋白质的标准序列中,而不出现在剪接变体中;(2)PTM和SAP可同时出现在由标准序列编码的蛋白质和剪接变体中;(3)所有修饰类型(AS、SAP和PTM)作为独立事件发生。蛋白质异构体的实验验证受到蛋白质组学技术分析灵敏度的限制。针对由单条染色体编码的蛋白质生成了一个钟形分布直方图,并估计了血浆、肝脏和HepG2细胞系中的拷贝数。所提出的元生物信息学方法可用于估计任何一组蛋白质编码基因的不同蛋白质异构体的数量。