Tang Rongying, Prosser Debra O, Love Donald R
Diagnostic Genetics, LabPLUS, Auckland City Hospital, P.O. Box 110031, Auckland 1148, New Zealand.
Adv Bioinformatics. 2016;2016:5614058. doi: 10.1155/2016/5614058. Epub 2016 May 24.
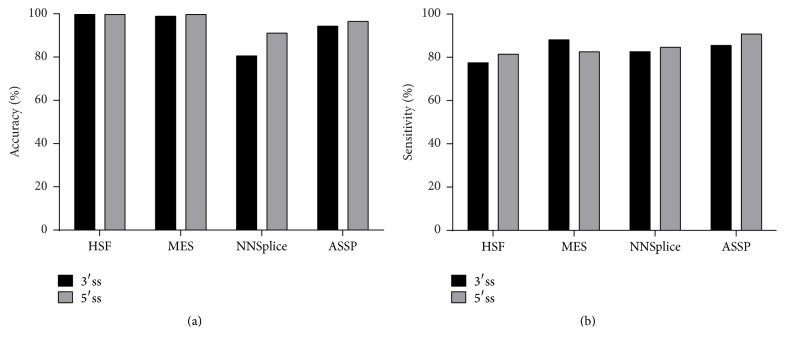
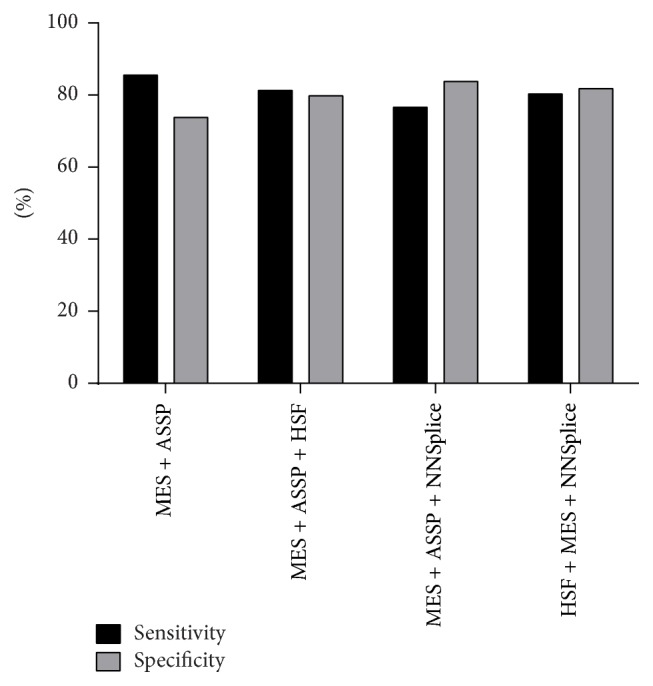
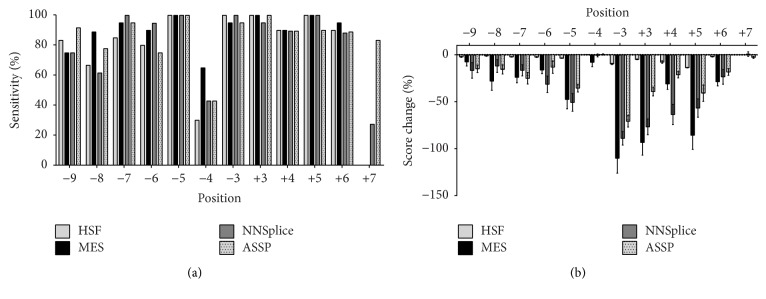
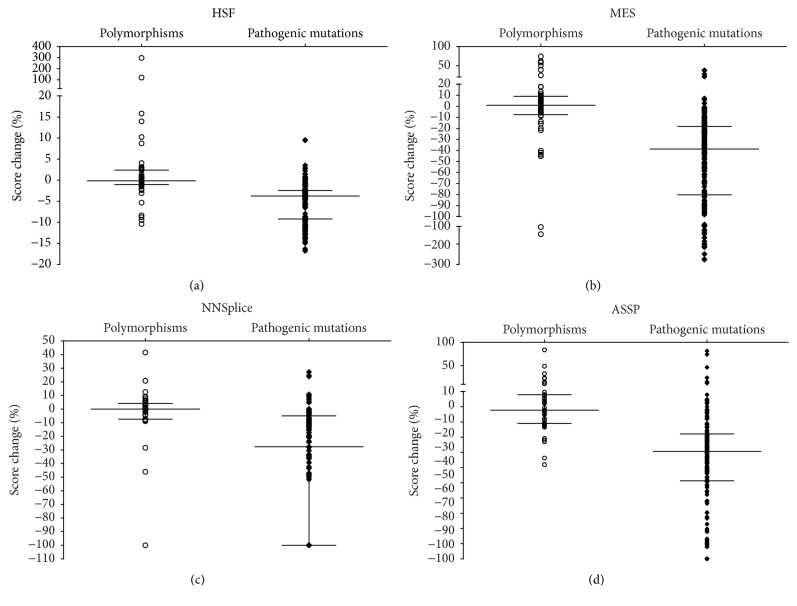
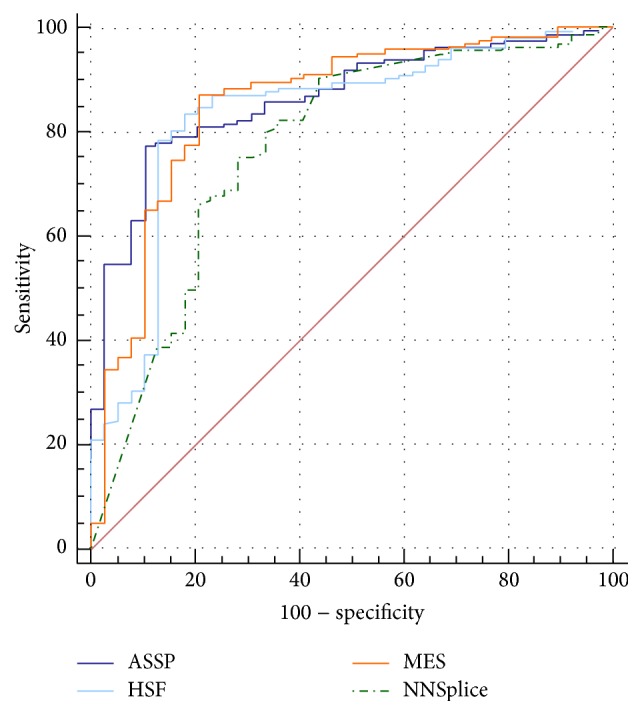
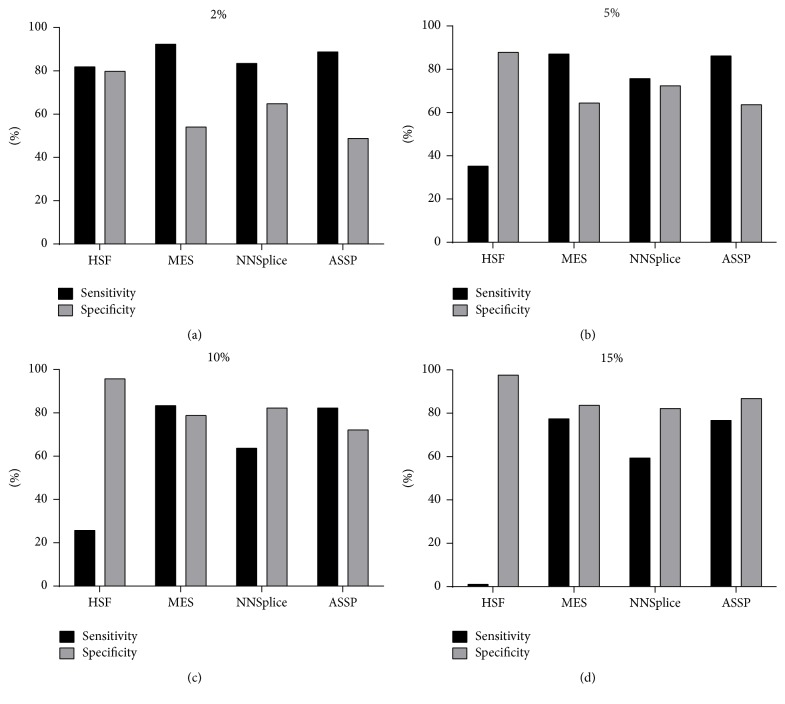
The increasing diagnostic use of gene sequencing has led to an expanding dataset of novel variants that lie within consensus splice junctions. The challenge for diagnostic laboratories is the evaluation of these variants in order to determine if they affect splicing or are merely benign. A common evaluation strategy is to use in silico analysis, and it is here that a number of programmes are available online; however, currently, there are no consensus guidelines on the selection of programmes or protocols to interpret the prediction results. Using a collection of 222 pathogenic mutations and 50 benign polymorphisms, we evaluated the sensitivity and specificity of four in silico programmes in predicting the effect of each variant on splicing. The programmes comprised Human Splice Finder (HSF), Max Entropy Scan (MES), NNSplice, and ASSP. The MES and ASSP programmes gave the highest performance based on Receiver Operator Curve analysis, with an optimal cut-off of score reduction of 10%. The study also showed that the sensitivity of prediction is affected by the level of conservation of individual positions, with in silico predictions for variants at positions -4 and +7 within consensus splice sites being largely uninformative.
基因测序在诊断中的应用日益增加,导致了位于共有剪接位点内的新型变异数据集不断扩大。诊断实验室面临的挑战是评估这些变异,以确定它们是否影响剪接,还是仅仅是良性的。一种常见的评估策略是使用计算机分析,目前有许多程序可在线获取;然而,目前在选择程序或方案以解释预测结果方面尚无共识指南。我们使用222个致病突变和50个良性多态性的集合,评估了四个计算机程序在预测每个变异对剪接的影响方面的敏感性和特异性。这些程序包括人类剪接查找器(HSF)、最大熵扫描(MES)、NNSplice和ASSP。根据受试者工作特征曲线分析,MES和ASSP程序表现最佳,最佳截断值为分数降低10%。该研究还表明,预测的敏感性受各个位置的保守程度影响,对于共有剪接位点内-4和+7位置的变异,计算机预测在很大程度上没有信息量。