Bryen Samantha J, Yuen Michaela, Joshi Himanshu, Dawes Ruebena, Zhang Katharine, Lu Jessica K, Jones Kristi J, Liang Christina, Wong Wui-Kwan, Peduto Anthony J, Waddell Leigh B, Evesson Frances J, Cooper Sandra T
Kids Neuroscience Centre, Kids Research, The Children's Hospital at Westmead, Locked Bag 4001, Westmead, NSW 2145, Australia.
Discipline of Child and Adolescent Health, Faculty of Medicine and Health, The University of Sydney, Locked Bag 4001, Westmead, NSW 2145, Australia.
HGG Adv. 2022 Jun 25;3(4):100125. doi: 10.1016/j.xhgg.2022.100125. eCollection 2022 Oct 13.
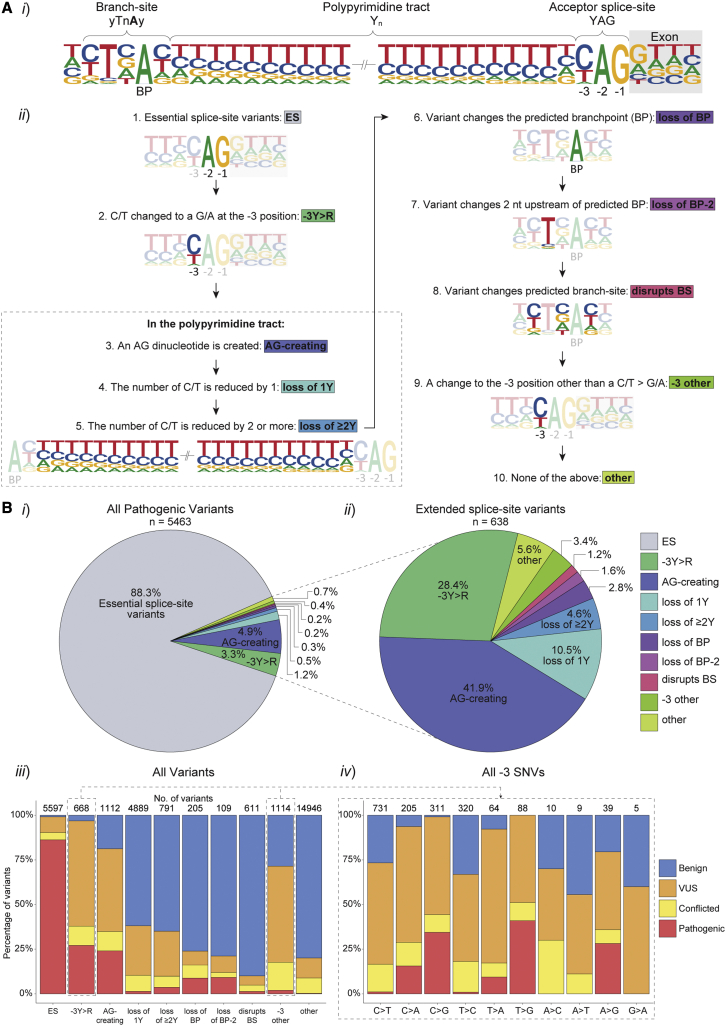
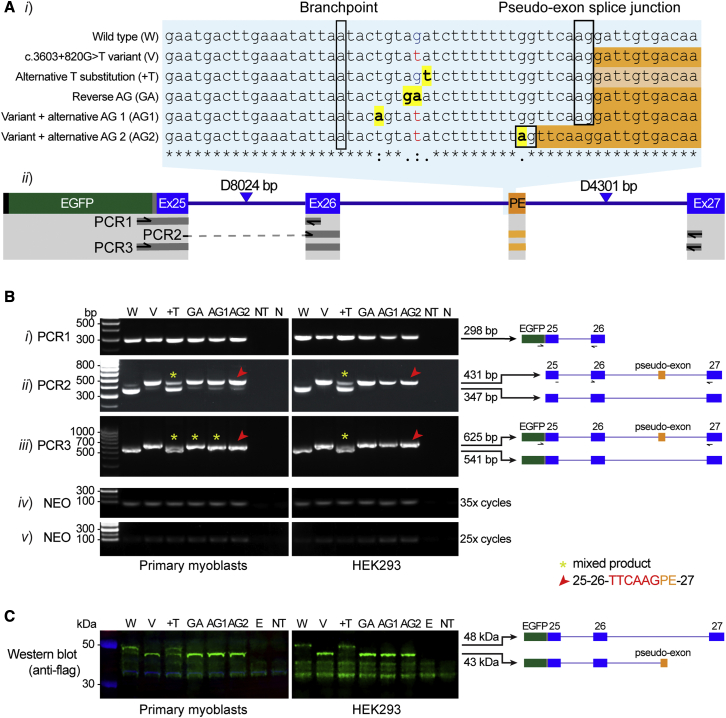
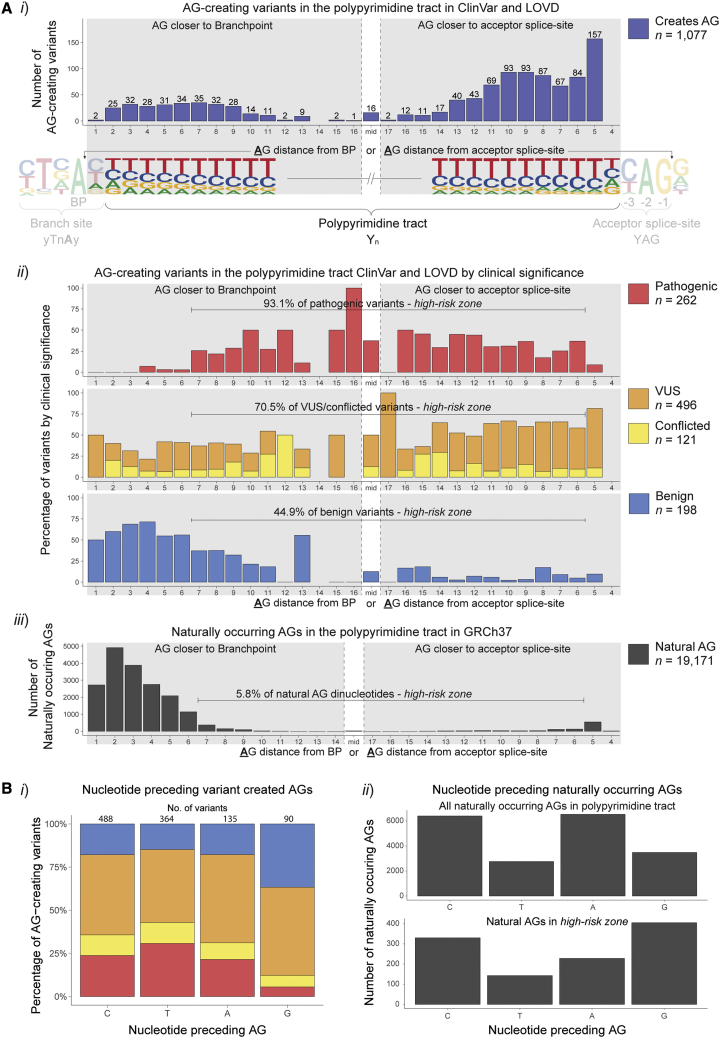
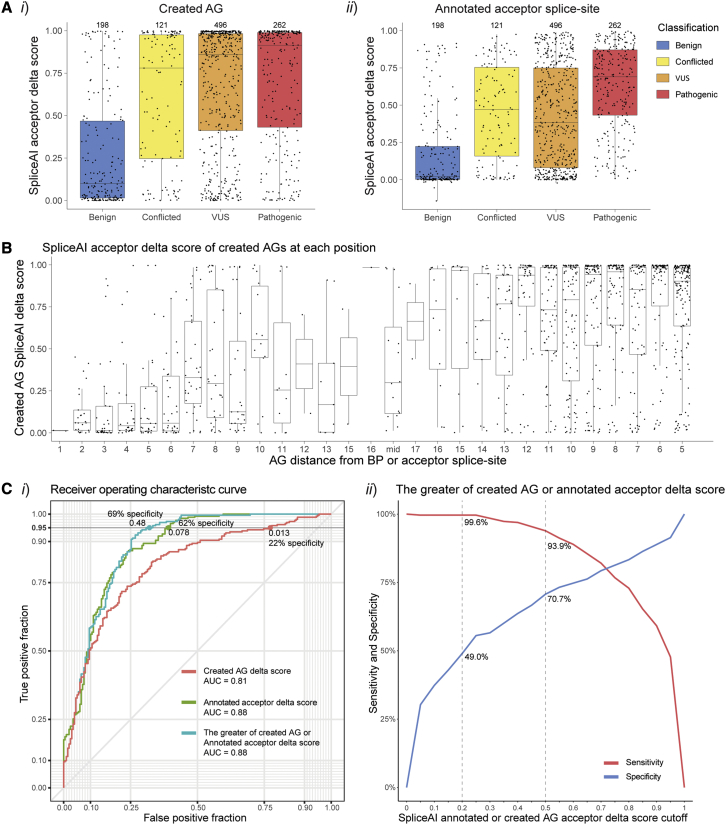
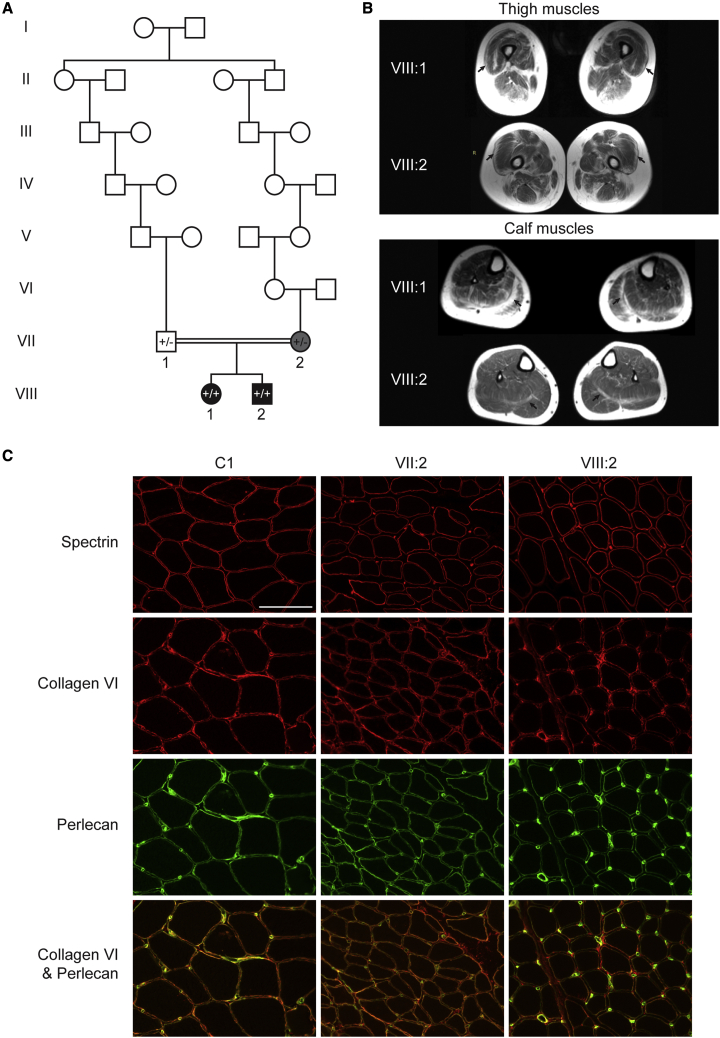
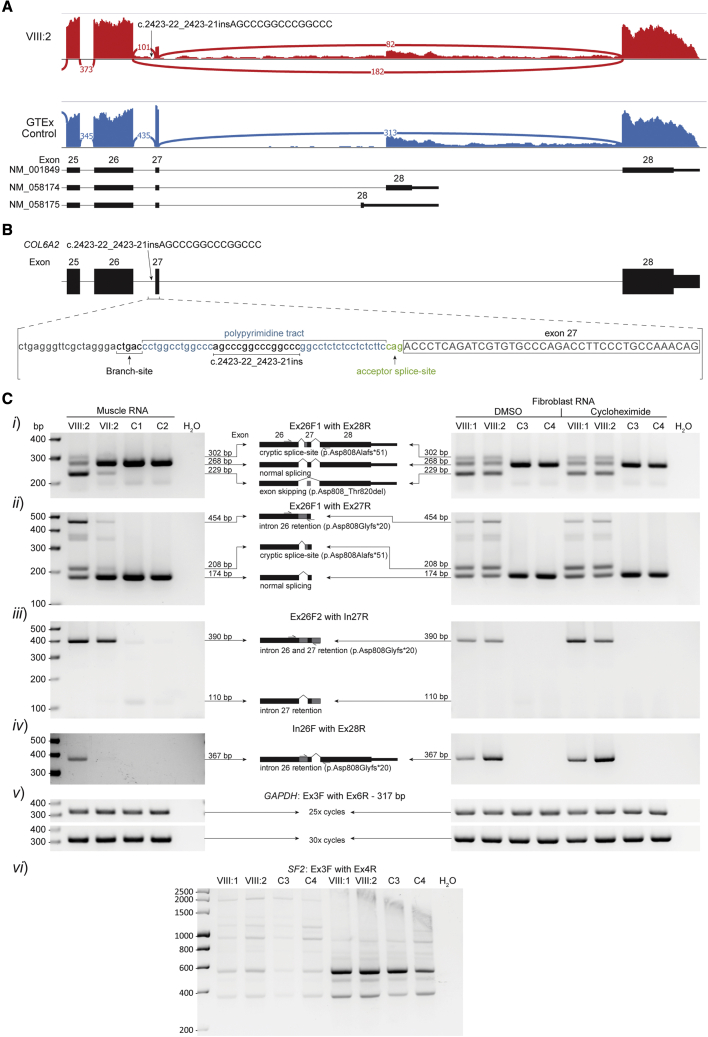
Predicting the pathogenicity of acceptor splice-site variants outside the essential AG is challenging, due to high sequence diversity of the extended splice-site region. Critical analysis of 24,445 intronic extended acceptor splice-site variants reported in ClinVar and the Leiden Open Variation Database (LOVD) demonstrates 41.9% of pathogenic variants create an AG dinucleotide between the predicted branchpoint and acceptor (AG-creating variants in the AG exclusion zone), 28.4% result in loss of a pyrimidine at the -3 position, and 15.1% result in loss of one or more pyrimidines in the polypyrimidine tract. Pathogenicity of AG-creating variants was highly influenced by their position. We define a for pathogenicity: > 6 nucleotides downstream of the predicted branchpoint and >5 nucleotides upstream from the acceptor, where 93.1% of pathogenic AG-creating variants arise and where naturally occurring AG dinucleotides are concordantly depleted (5.8% of natural AGs). SpliceAI effectively predicts pathogenicity of AG-creating variants, achieving 95% sensitivity and 69% specificity. We highlight clinical examples showing contrasting mechanisms for mis-splicing arising from AG variants: (1) cryptic acceptor created; (2) splicing silencer created: an introduced AG silences the acceptor, resulting in exon skipping, intron retention, and/or use of an alternative existing cryptic acceptor; and (3) splicing silencer disrupted: loss of a deep intronic AG activates inclusion of a pseudo-exon. In conclusion, we establish AG-creating variants as a common class of pathogenic extended acceptor variant and outline factors conferring critical risk for mis-splicing for AG-creating variants in the AG exclusion zone, between the branchpoint and acceptor.
预测基本AG以外的受体剪接位点变异的致病性具有挑战性,因为扩展剪接位点区域的序列多样性很高。对ClinVar和莱顿开放变异数据库(LOVD)中报告的24445个内含子扩展受体剪接位点变异进行的批判性分析表明,41.9%的致病变异在预测的分支点和受体之间产生了AG二核苷酸(AG排除区内的AG产生变异),28.4%导致-3位置的嘧啶缺失,15.1%导致多嘧啶序列中一个或多个嘧啶缺失。AG产生变异的致病性受其位置的影响很大。我们定义了一个致病性区域:在预测分支点下游>6个核苷酸且在受体上游>5个核苷酸处,93.1%的致病性AG产生变异出现在该区域,且天然存在的AG二核苷酸在此区域一致减少(天然AG的5.8%)。SpliceAI有效地预测了AG产生变异的致病性,灵敏度达到95%,特异性达到69%。我们重点介绍了临床实例,展示了AG变异导致错配剪接的不同机制:(1)产生隐蔽受体;(2)产生剪接沉默子:引入的AG使受体沉默,导致外显子跳跃、内含子保留和/或使用替代的现有隐蔽受体;(3)剪接沉默子破坏:内含子深处的AG缺失激活了假外显子的包含。总之,我们将AG产生变异确定为一类常见的致病性扩展受体变异,并概述了在分支点和受体之间的AG排除区内,AG产生变异导致错配剪接的关键风险因素。