Bergmann Ewa A, Chen Bo-Juen, Arora Kanika, Vacic Vladimir, Zody Michael C
New York Genome Center, New York, NY 10013, USA.
Bioinformatics. 2016 Oct 15;32(20):3196-3198. doi: 10.1093/bioinformatics/btw389. Epub 2016 Jun 26.
Sequencing of matched tumor and normal samples is the standard study design for reliable detection of somatic alterations. However, even very low levels of cross-sample contamination significantly impact calling of somatic mutations, because contaminant germline variants can be incorrectly interpreted as somatic. There are currently no sequence-only based methods that reliably estimate contamination levels in tumor samples, which frequently display copy number changes. As a solution, we developed Conpair, a tool for detection of sample swaps and cross-individual contamination in whole-genome and whole-exome tumor-normal sequencing experiments.
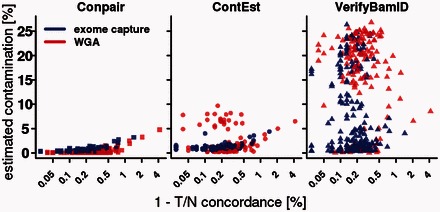
On a ladder of in silico contaminated samples, we demonstrated that Conpair reliably measures contamination levels as low as 0.1%, even in presence of copy number changes. We also estimated contamination levels in glioblastoma WGS and WXS tumor-normal datasets from TCGA and showed that they strongly correlate with tumor-normal concordance, as well as with the number of germline variants called as somatic by several widely-used somatic callers.
The method is available at: https://github.com/nygenome/conpair CONTACT: egrabowska@gmail.com or mczody@nygenome.orgSupplementary information: Supplementary data are available at Bioinformatics online.
对匹配的肿瘤样本和正常样本进行测序是可靠检测体细胞改变的标准研究设计。然而,即使是极低水平的跨样本污染也会显著影响体细胞突变的检测,因为污染的种系变异可能会被错误地解释为体细胞变异。目前还没有基于序列的方法能够可靠地估计肿瘤样本中的污染水平,而肿瘤样本经常会出现拷贝数变化。作为一种解决方案,我们开发了Conpair,这是一种用于在全基因组和全外显子肿瘤-正常测序实验中检测样本交换和个体间污染的工具。
在一系列计算机模拟污染样本上,我们证明了Conpair即使在存在拷贝数变化的情况下也能可靠地测量低至0.1%的污染水平。我们还估计了来自TCGA的胶质母细胞瘤全基因组测序(WGS)和全外显子测序(WXS)肿瘤-正常数据集的污染水平,结果表明它们与肿瘤-正常一致性以及几种广泛使用的体细胞变异检测工具误判为体细胞变异的种系变异数量密切相关。
该方法可在以下网址获取:https://github.com/nygenome/conpair 联系方式:egrabowska@gmail.com 或 mczody@nygenome.org 补充信息:补充数据可在《生物信息学》在线获取。