Calus Mario P L, Bouwman Aniek C, Schrooten Chris, Veerkamp Roel F
Animal Breeding and Genomics Centre, Wageningen UR Livestock Research, PO Box 338, 6700 AH, Wageningen, The Netherlands.
CRV BV, 6800 AL, Arnhem, The Netherlands.
Genet Sel Evol. 2016 Jun 29;48(1):49. doi: 10.1186/s12711-016-0225-x.
Use of whole-genome sequence data is expected to increase persistency of genomic prediction across generations and breeds but affects model performance and requires increased computing time. In this study, we investigated whether the split-and-merge Bayesian stochastic search variable selection (BSSVS) model could overcome these issues. BSSVS is performed first on subsets of sequence-based variants and then on a merged dataset containing variants selected in the first step.
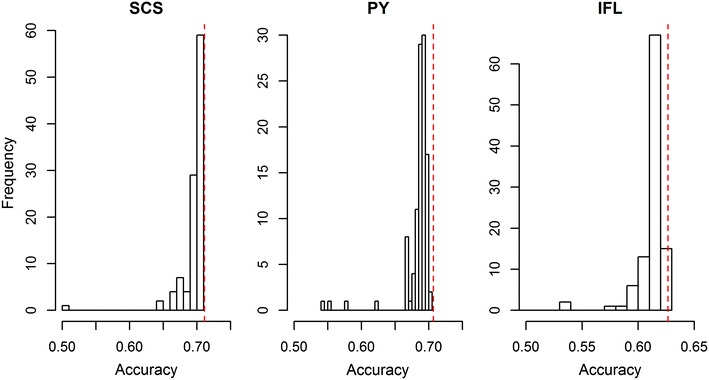
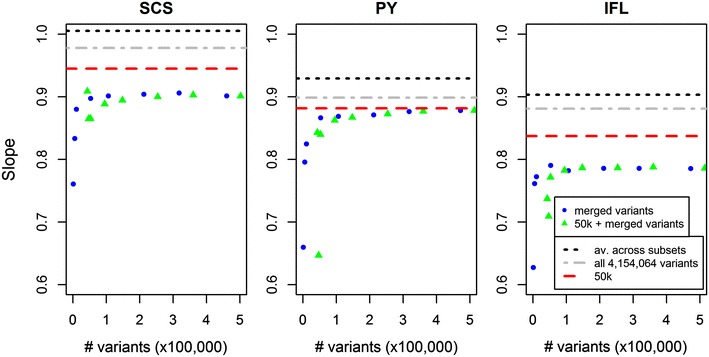
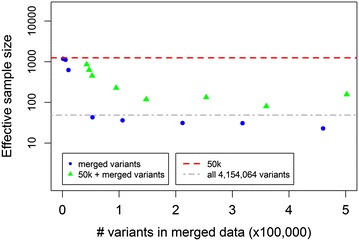
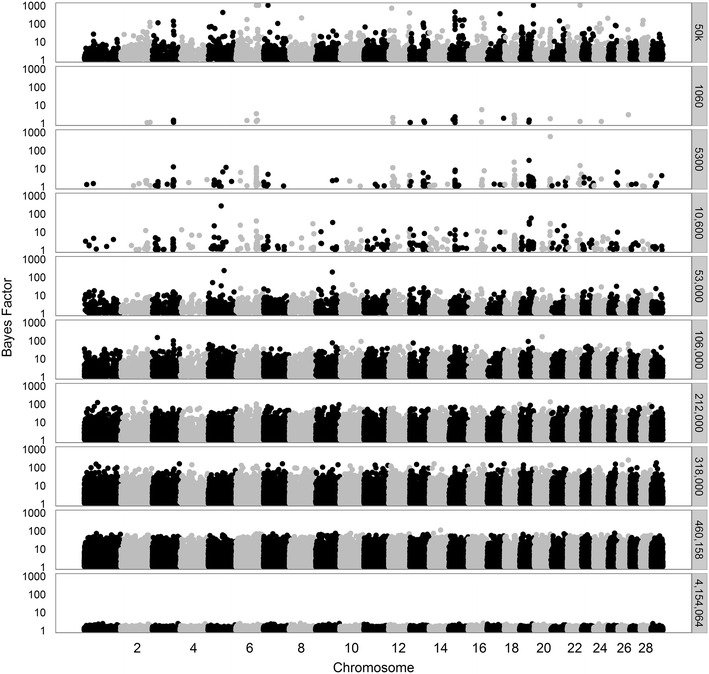
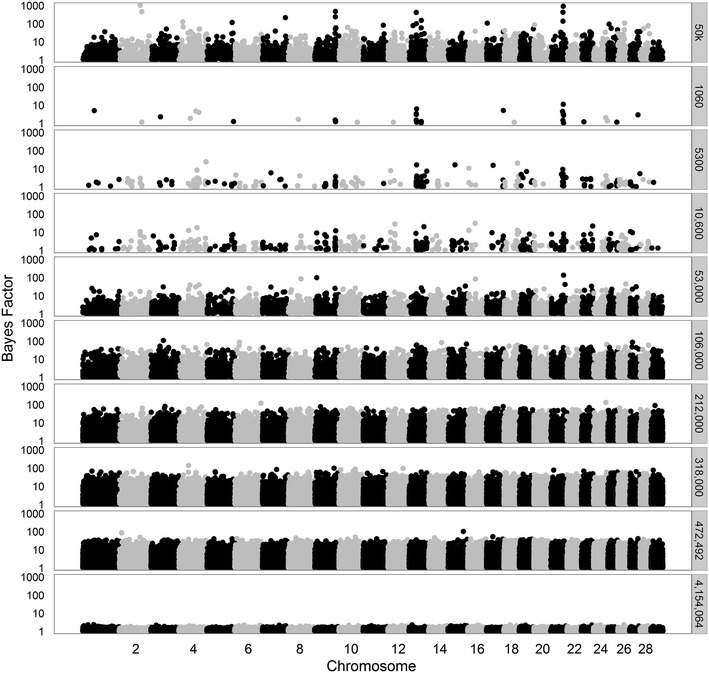
We used a dataset that included 4,154,064 variants after editing and de-regressed proofs for 3415 reference and 2138 validation bulls for somatic cell score, protein yield and interval first to last insemination. In the first step, BSSVS was performed on 106 subsets each containing ~39,189 variants. In the second step, 1060 up to 472,492 variants, selected from the first step, were included to estimate the accuracy of genomic prediction. Accuracies were at best equal to those achieved with the commonly used Bovine 50k-SNP chip, although the number of variants within a few well-known quantitative trait loci regions was considerably enriched. When variant selection and the final genomic prediction were performed on the same data, predictions were biased. Predictions computed as the average of the predictions computed for each subset achieved the highest accuracies, i.e. 0.5 to 1.1 % higher than the accuracies obtained with the 50k-SNP chip, and yielded the least biased predictions. Finally, the accuracy of genomic predictions obtained when all sequence-based variants were included was similar or up to 1.4 % lower compared to that based on the average predictions across the subsets. By applying parallelization, the split-and-merge procedure was completed in 5 days, while the standard analysis including all sequence-based variants took more than three months.
The split-and-merge approach splits one large computational task into many much smaller ones, which allows the use of parallel processing and thus efficient genomic prediction based on whole-genome sequence data. The split-and-merge approach did not improve prediction accuracy, probably because we used data on a single breed for which relationships between individuals were high. Nevertheless, the split-and-merge approach may have potential for applications on data from multiple breeds.
全基因组序列数据的使用有望提高基因组预测在各代和各品种间的持续性,但会影响模型性能且需要增加计算时间。在本研究中,我们调查了拆分合并贝叶斯随机搜索变量选择(BSSVS)模型是否能克服这些问题。BSSVS首先在基于序列的变异子集上进行,然后在包含第一步中选择的变异的合并数据集上进行。
我们使用了一个数据集,该数据集在对3415头参考公牛和2138头验证公牛的体细胞评分、蛋白质产量以及首次输精到最后一次输精的间隔进行编辑和去回归证明后,包含4,154,064个变异。在第一步中,BSSVS在106个子集上进行,每个子集包含约39,189个变异。在第二步中,纳入从第一步中选择的1060至472,492个变异,以估计基因组预测的准确性。尽管一些知名数量性状基因座区域内的变异数量大幅增加,但准确性最高仅与使用常用的牛50k-SNP芯片时相当。当在相同数据上进行变异选择和最终基因组预测时,预测存在偏差。将每个子集计算的预测值求平均得到的预测结果准确性最高,即比使用50k-SNP芯片获得的准确性高0.5%至1.1%,且偏差最小。最后,与基于各子集平均预测结果相比,纳入所有基于序列的变异时获得的基因组预测准确性相似或低至1.4%。通过应用并行化,拆分合并过程在5天内完成,而包括所有基于序列变异的标准分析则耗时三个多月。
拆分合并方法将一个大型计算任务拆分为许多小得多的任务,这使得能够使用并行处理,从而基于全基因组序列数据进行高效的基因组预测。拆分合并方法并未提高预测准确性,可能是因为我们使用的是单个品种的数据,个体间亲缘关系较高。尽管如此,拆分合并方法可能在多品种数据的应用中具有潜力。