Chella Krishnan Karthickeyan, Mukundan Santhosh, Alagarsamy Jeyashree, Hur Junguk, Nookala Suba, Siemens Nikolai, Svensson Mattias, Hyldegaard Ole, Norrby-Teglund Anna, Kotb Malak
Department of Molecular Genetics, Biochemistry and Microbiology, College of Medicine, University of Cincinnati, Cincinnati, Ohio, United States of America.
Department of Biomedical Sciences, School of Medicine and Health Sciences, University of North Dakota, Grand Forks, North Dakota, United States of America.
PLoS Pathog. 2016 Jul 11;12(7):e1005732. doi: 10.1371/journal.ppat.1005732. eCollection 2016 Jul.
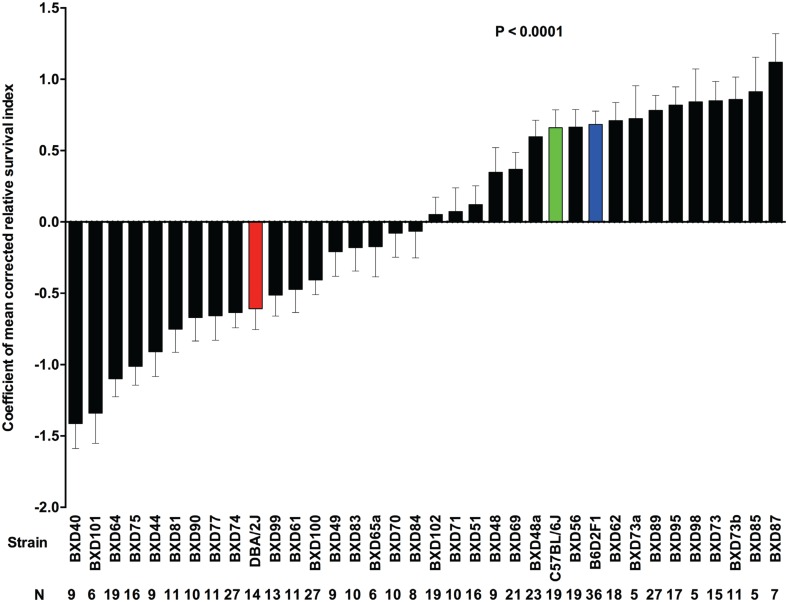
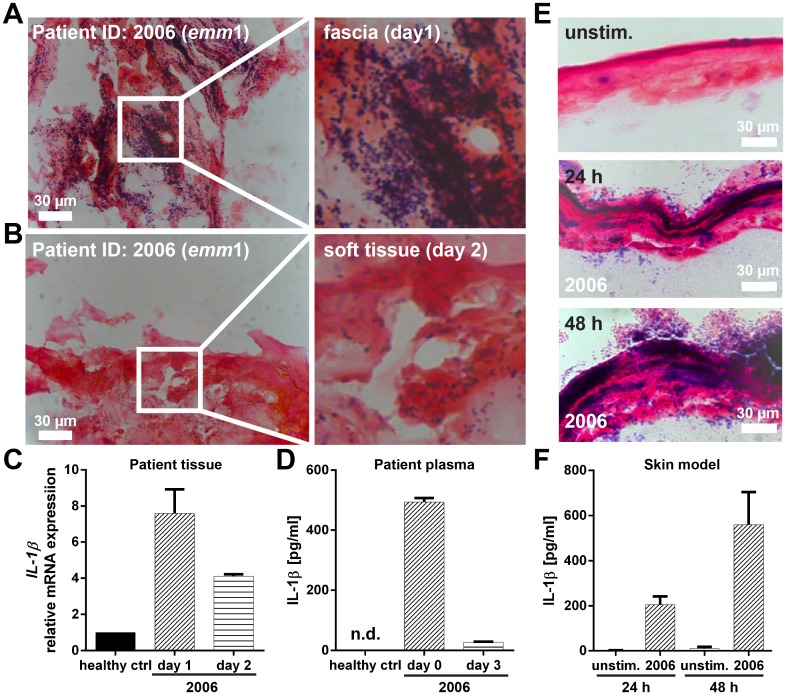
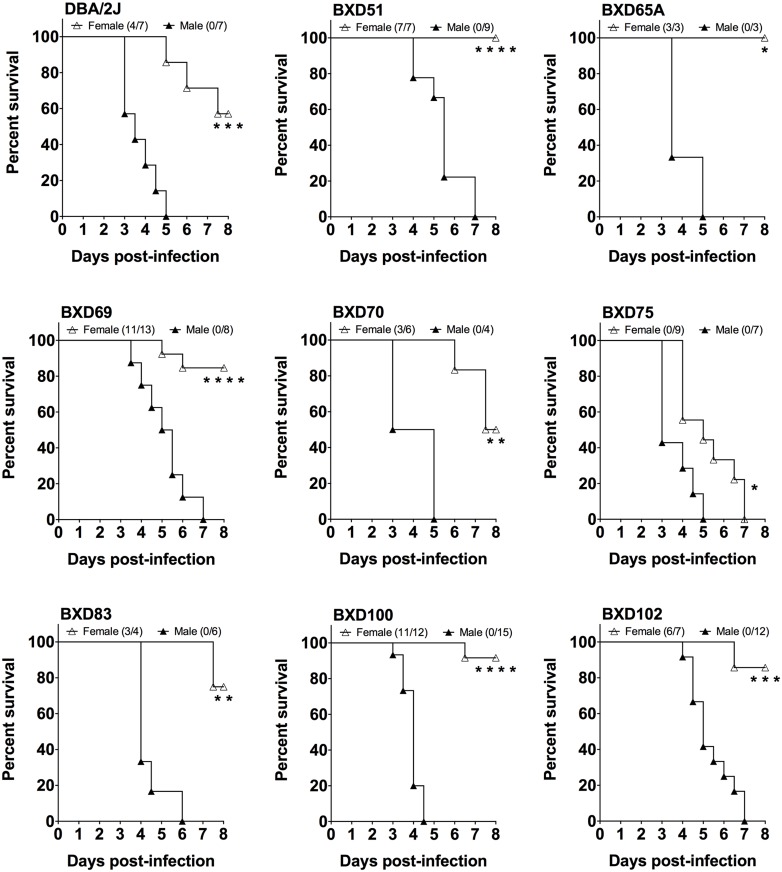
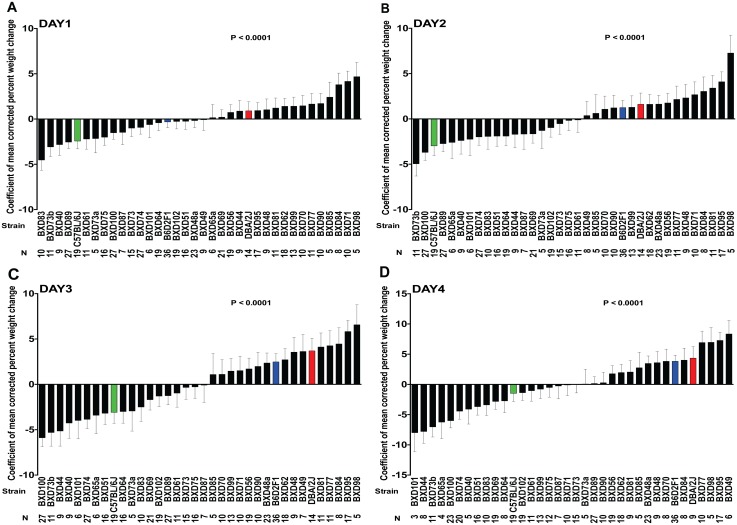
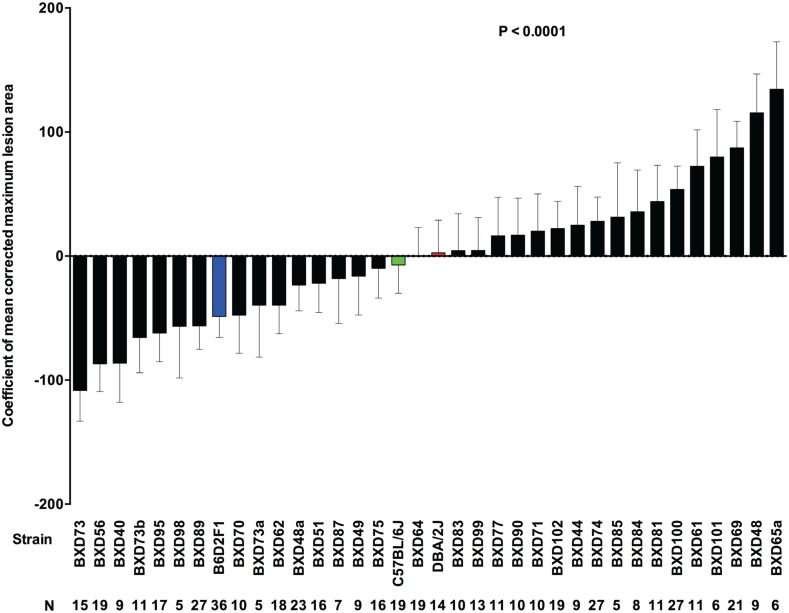
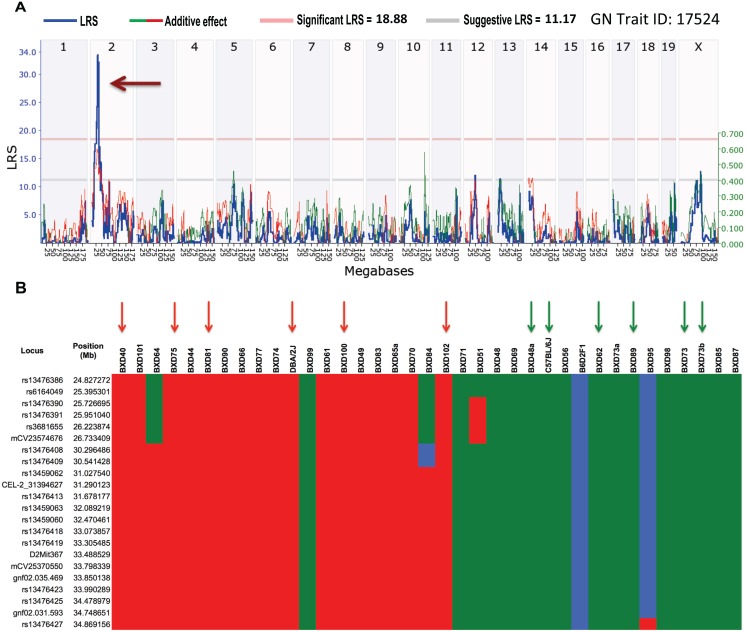
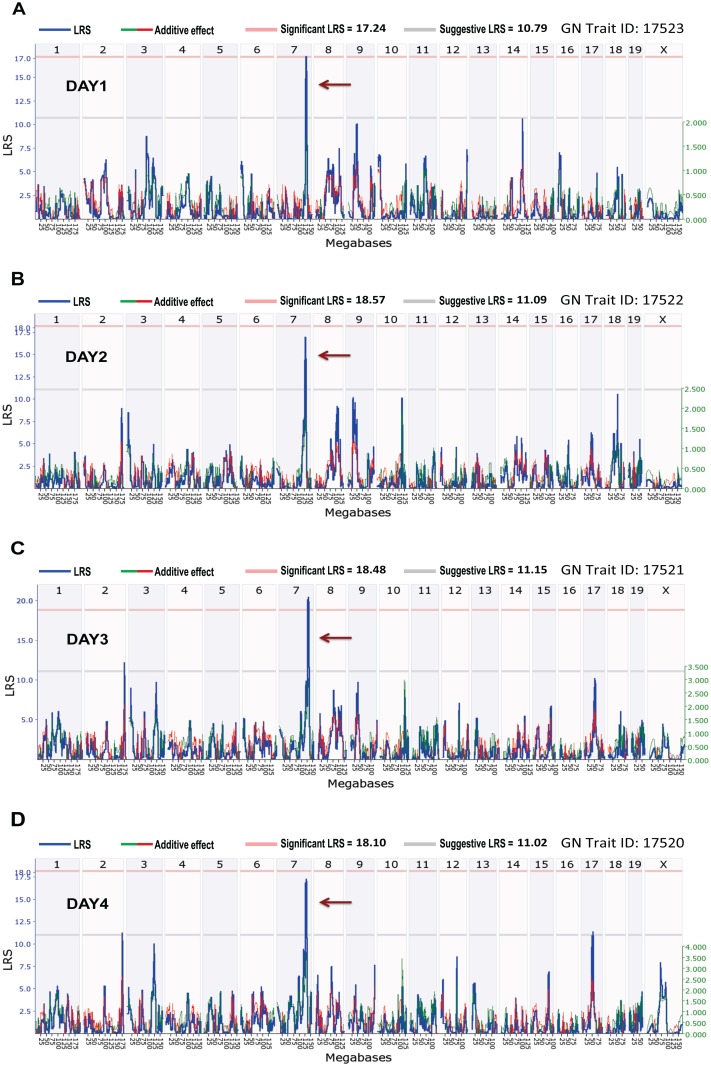
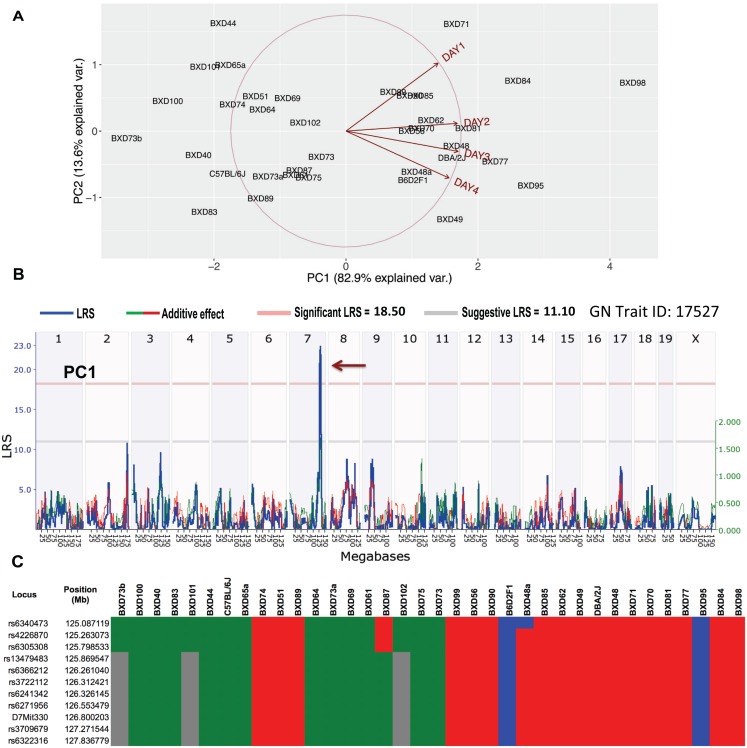
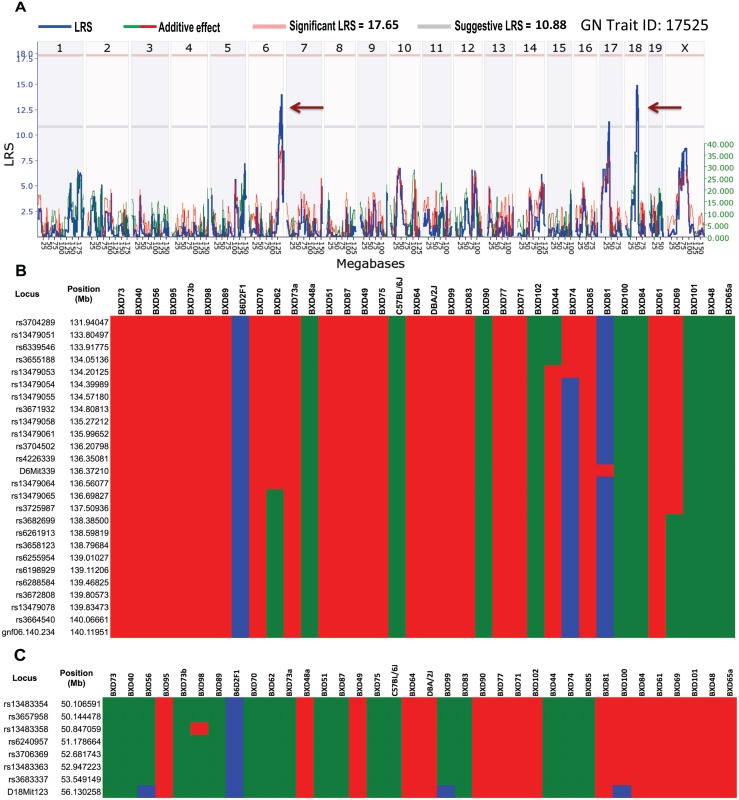
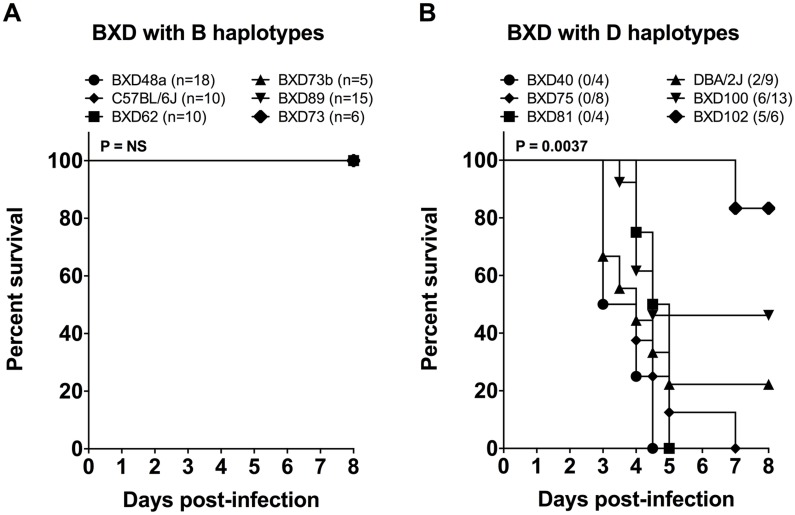
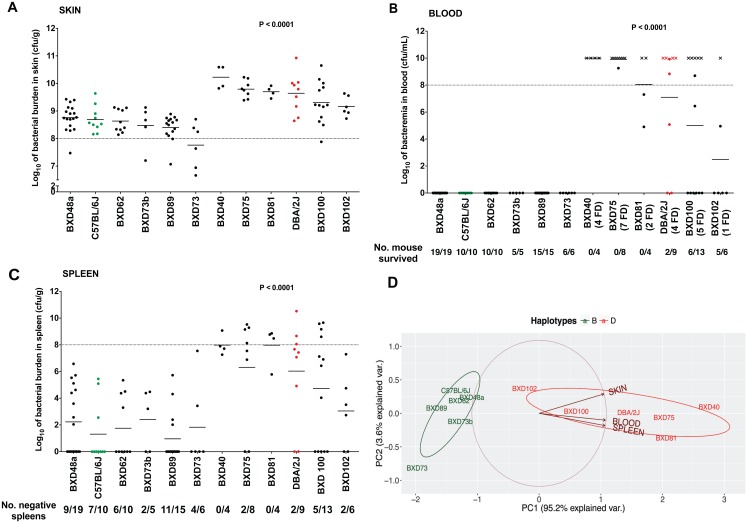
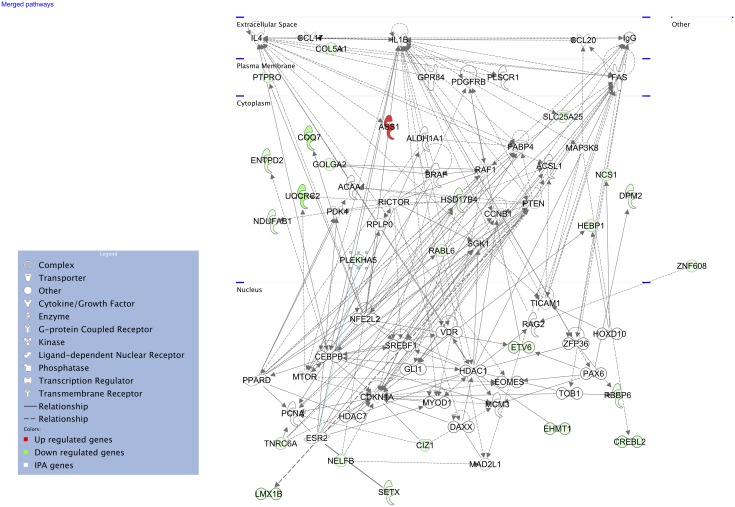
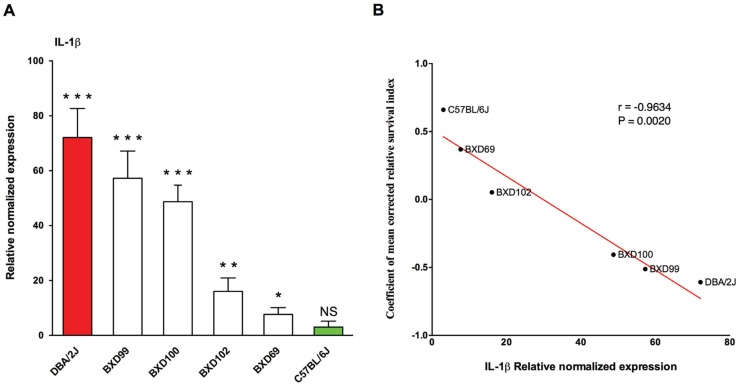
Host genetic variations play an important role in several pathogenic diseases, and we have previously provided strong evidences that these genetic variations contribute significantly to differences in susceptibility and clinical outcomes of invasive Group A Streptococcus (GAS) infections, including sepsis and necrotizing soft tissue infections (NSTIs). Our initial studies with conventional mouse strains revealed that host genetic variations and sex differences play an important role in orchestrating the severity, susceptibility and outcomes of NSTIs. To understand the complex genetic architecture of NSTIs, we utilized an unbiased, forward systems genetics approach in an advanced recombinant inbred (ARI) panel of mouse strains (BXD). Through this approach, we uncovered interactions between host genetics, and other non-genetic cofactors including sex, age and body weight in determining susceptibility to NSTIs. We mapped three NSTIs-associated phenotypic traits (i.e., survival, percent weight change, and lesion size) to underlying host genetic variations by using the WebQTL tool, and identified four NSTIs-associated quantitative genetic loci (QTL) for survival on mouse chromosome (Chr) 2, for weight change on Chr 7, and for lesion size on Chr 6 and 18 respectively. These QTL harbor several polymorphic genes. Identification of multiple QTL highlighted the complexity of the host-pathogen interactions involved in NSTI pathogenesis. We then analyzed and rank-ordered host candidate genes in these QTL by using the QTLminer tool and then developed a list of 375 candidate genes on the basis of annotation data and biological relevance to NSTIs. Further differential expression analyses revealed 125 genes to be significantly differentially regulated in susceptible strains compared to their uninfected controls. Several of these genes are involved in innate immunity, inflammatory response, cell growth, development and proliferation, and apoptosis. Additional network analyses using ingenuity pathway analysis (IPA) of these 125 genes revealed interleukin-1 beta network as key network involved in modulating the differential susceptibility to GAS NSTIs.
宿主基因变异在多种致病疾病中发挥着重要作用,我们之前已提供了有力证据,表明这些基因变异对侵袭性A组链球菌(GAS)感染(包括败血症和坏死性软组织感染(NSTIs))的易感性差异和临床结局有显著影响。我们对传统小鼠品系的初步研究表明,宿主基因变异和性别差异在协调NSTIs的严重程度、易感性和结局方面起着重要作用。为了了解NSTIs复杂的遗传结构,我们在一组先进的重组近交(ARI)小鼠品系(BXD)中采用了无偏向前瞻性系统遗传学方法。通过这种方法,我们发现了宿主遗传学与其他非遗传辅助因素(包括性别、年龄和体重)之间在决定对NSTIs易感性方面的相互作用。我们使用WebQTL工具将三个与NSTIs相关的表型性状(即存活率、体重变化百分比和病变大小)映射到潜在的宿主基因变异上,并分别在小鼠染色体(Chr)2上鉴定出四个与NSTIs相关的存活数量性状基因座(QTL),在Chr 7上鉴定出与体重变化相关的QTL,在Chr 6和18上分别鉴定出与病变大小相关的QTL。这些QTL包含多个多态性基因。多个QTL的鉴定突出了NSTI发病机制中宿主 - 病原体相互作用的复杂性。然后,我们使用QTLminer工具对这些QTL中的宿主候选基因进行分析并排序,随后根据注释数据和与NSTIs的生物学相关性列出了375个候选基因清单。进一步的差异表达分析显示,与未感染对照相比,易感品系中有125个基因受到显著差异调节。其中一些基因参与先天免疫、炎症反应、细胞生长、发育和增殖以及细胞凋亡。使用 Ingenuity Pathway Analysis(IPA)对这125个基因进行的额外网络分析表明,白细胞介素 - 1β网络是参与调节对GAS NSTIs差异易感性的关键网络。