Kludas Jana, Arvas Mikko, Castillo Sandra, Pakula Tiina, Oja Merja, Brouard Céline, Jäntti Jussi, Penttilä Merja, Rousu Juho
Helsinki Institute for Information Technology HIIT, Department of Computer Science, Aalto University, Espoo, Finland.
VTT Technical Research Centre of Finland, Espoo, Finland.
PLoS One. 2016 Jul 21;11(7):e0159302. doi: 10.1371/journal.pone.0159302. eCollection 2016.
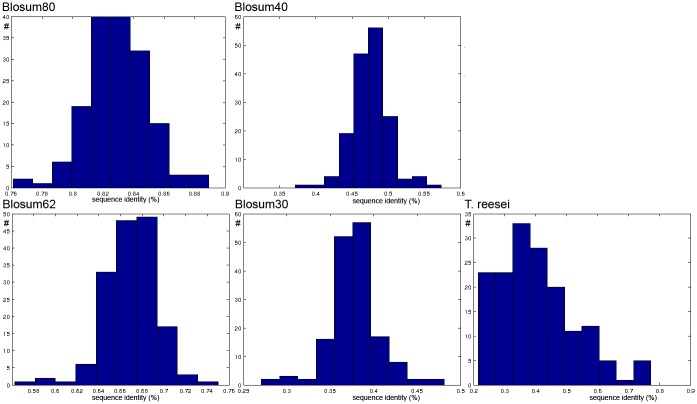
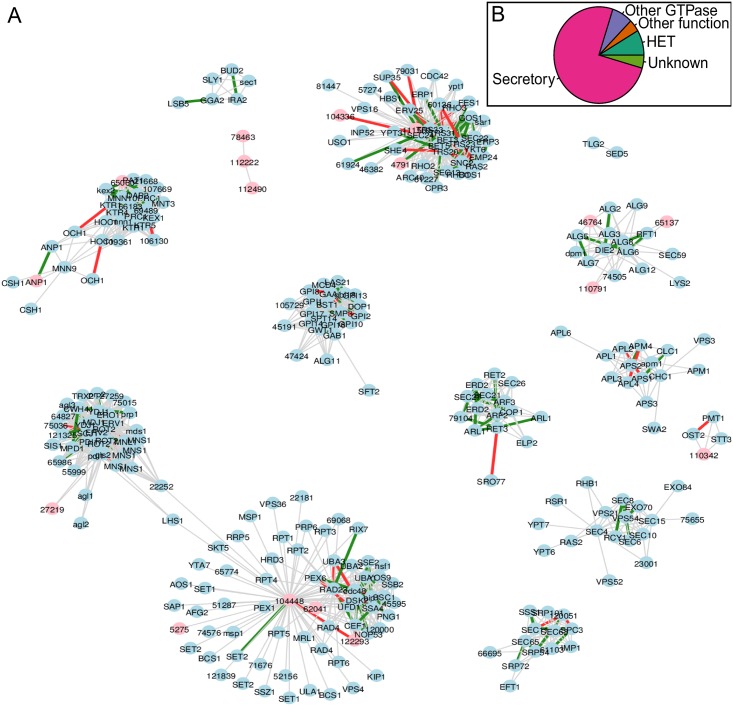
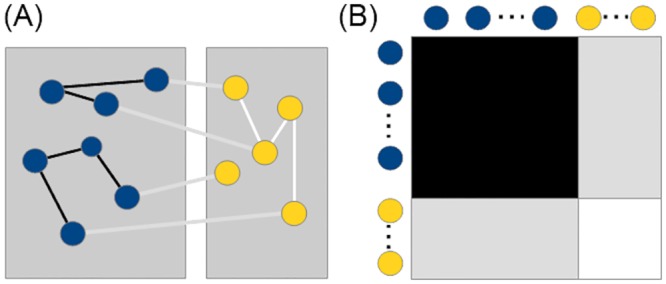
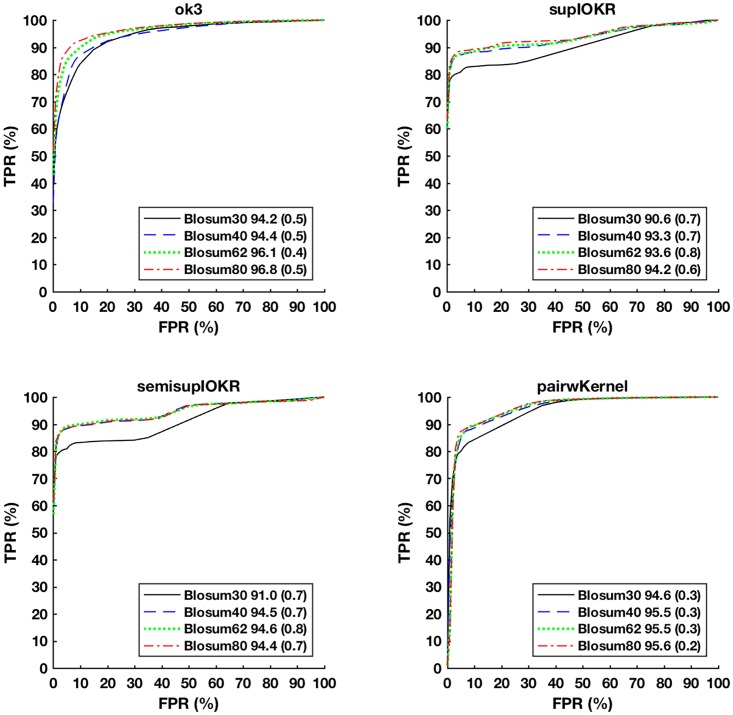
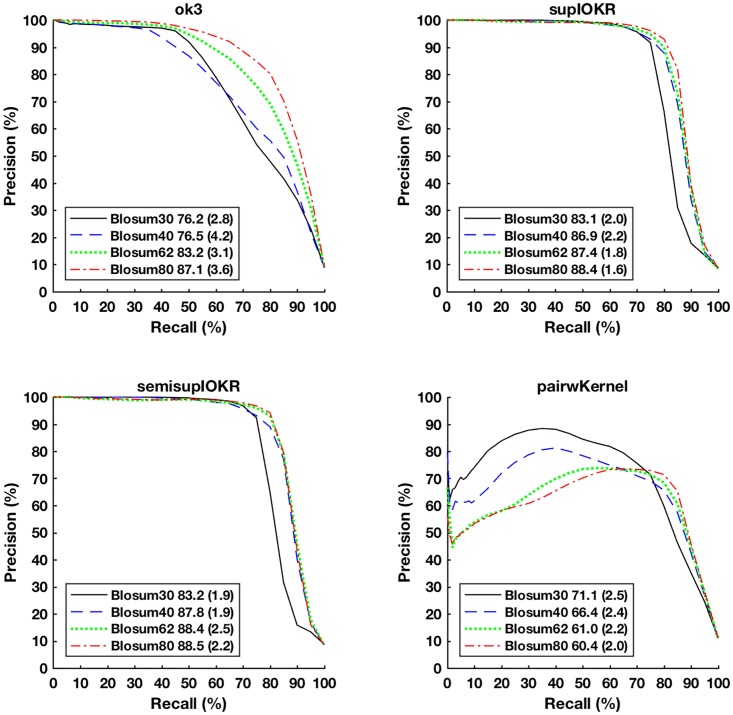
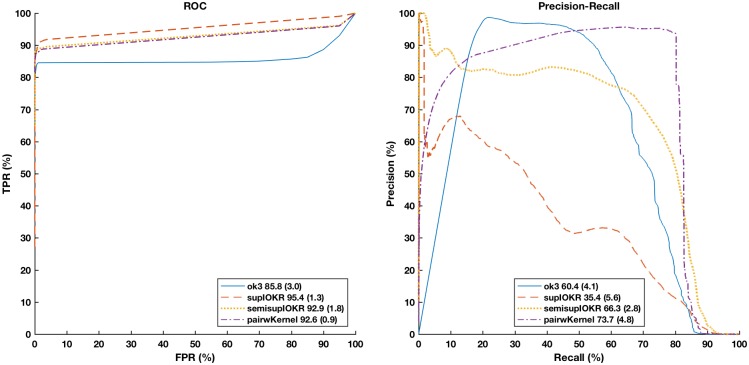
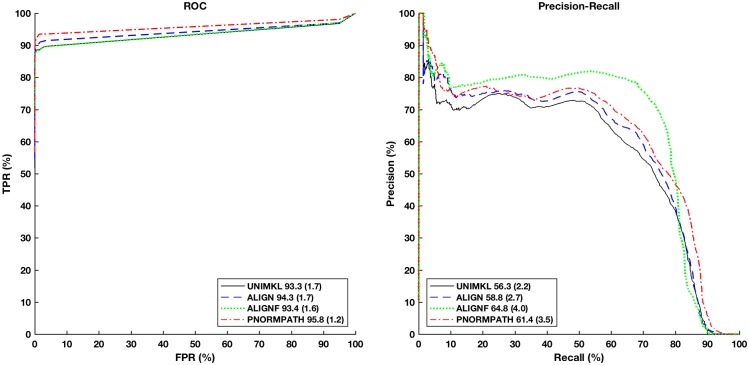
In this paper we apply machine learning methods for predicting protein interactions in fungal secretion pathways. We assume an inter-species transfer setting, where training data is obtained from a single species and the objective is to predict protein interactions in other, related species. In our methodology, we combine several state of the art machine learning approaches, namely, multiple kernel learning (MKL), pairwise kernels and kernelized structured output prediction in the supervised graph inference framework. For MKL, we apply recently proposed centered kernel alignment and p-norm path following approaches to integrate several feature sets describing the proteins, demonstrating improved performance. For graph inference, we apply input-output kernel regression (IOKR) in supervised and semi-supervised modes as well as output kernel trees (OK3). In our experiments simulating increasing genetic distance, Input-Output Kernel Regression proved to be the most robust prediction approach. We also show that the MKL approaches improve the predictions compared to uniform combination of the kernels. We evaluate the methods on the task of predicting protein-protein-interactions in the secretion pathways in fungi, S.cerevisiae, baker's yeast, being the source, T. reesei being the target of the inter-species transfer learning. We identify completely novel candidate secretion proteins conserved in filamentous fungi. These proteins could contribute to their unique secretion capabilities.
在本文中,我们应用机器学习方法来预测真菌分泌途径中的蛋白质相互作用。我们假设一种跨物种转移的情况,即训练数据从单个物种获取,目标是预测其他相关物种中的蛋白质相互作用。在我们的方法中,我们在监督图推理框架中结合了几种先进的机器学习方法,即多核学习(MKL)、成对核和核结构化输出预测。对于MKL,我们应用最近提出的中心核对齐和p范数路径跟踪方法来整合描述蛋白质的几个特征集,证明性能有所提高。对于图推理,我们在监督和半监督模式下应用输入-输出核回归(IOKR)以及输出核树(OK3)。在我们模拟遗传距离增加的实验中,输入-输出核回归被证明是最稳健的预测方法。我们还表明,与核的均匀组合相比,MKL方法改进了预测。我们在预测真菌分泌途径中蛋白质-蛋白质相互作用的任务上评估这些方法,酿酒酵母作为跨物种转移学习的源物种,里氏木霉作为目标物种。我们鉴定出在丝状真菌中保守的全新候选分泌蛋白。这些蛋白质可能有助于它们独特的分泌能力。