Gerasimidis Konstantinos, Bertz Martin, Quince Christopher, Brunner Katja, Bruce Alanna, Combet Emilie, Calus Szymon, Loman Nick, Ijaz Umer Zeeshan
Human Nutrition, School of Medicine, College of Medical, Veterinary and Life Sciences, Glasgow Royal Infirmary, University of Glasgow, Glasgow, UK.
Warwick Medical School, University of Warwick, Warwick, UK.
BMC Res Notes. 2016 Jul 26;9:365. doi: 10.1186/s13104-016-2171-7.
The effect that traditional and modern DNA extraction methods have on applications to study the role of gut microbiota in health and disease is a topic of current interest. Genomic DNA was extracted from three faecal samples and one probiotic capsule using three popular methods; chaotropic (CHAO) method, phenol/chloroform (PHEC) extraction, proprietary kit (QIAG). The performance of each of these methods on DNA yield and quality, microbiota composition using quantitative PCR, deep sequencing of the 16S rRNA gene, and sequencing analysis pipeline was evaluated.
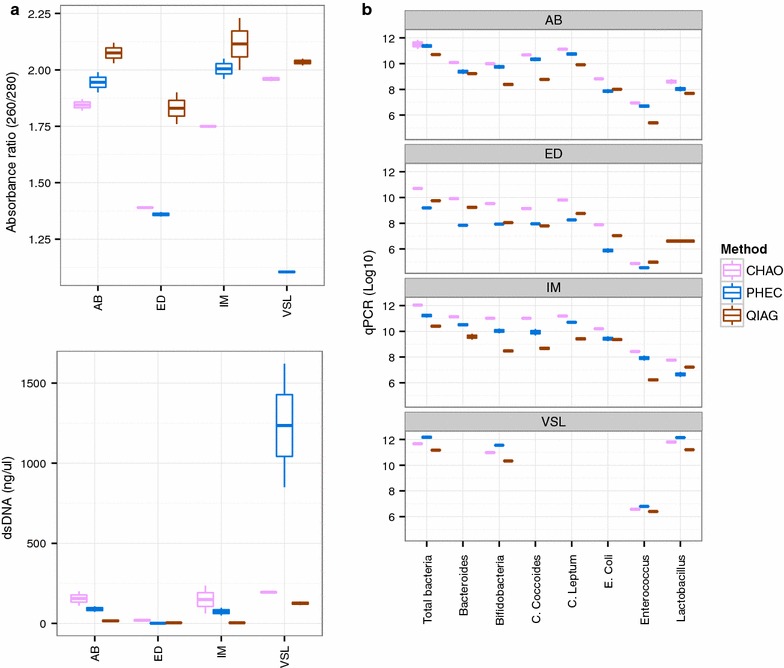
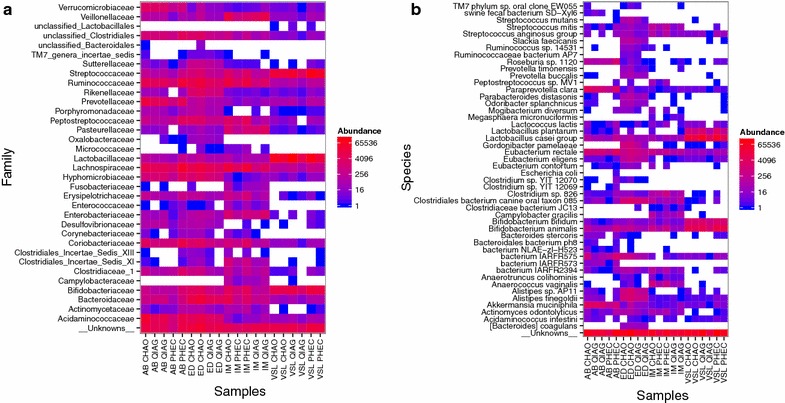
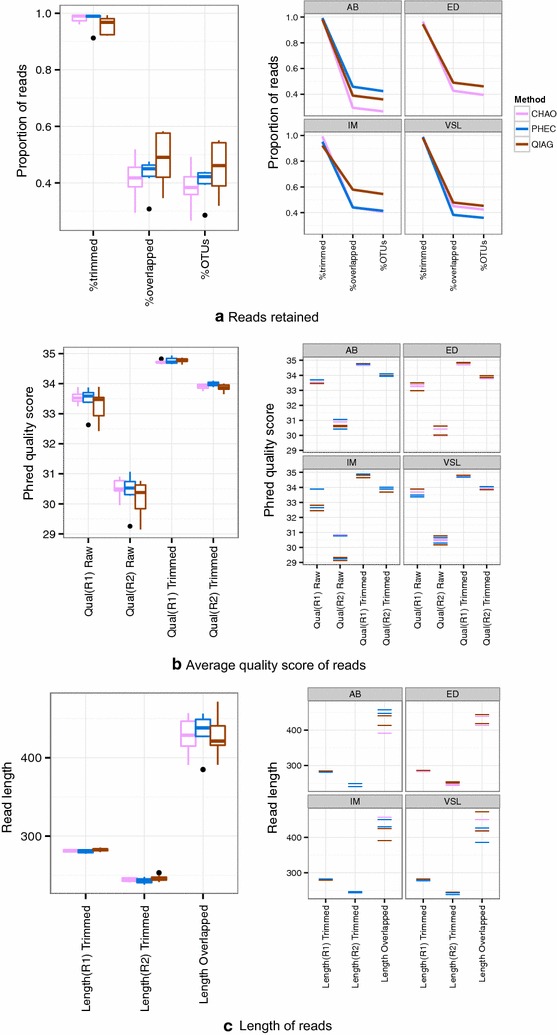
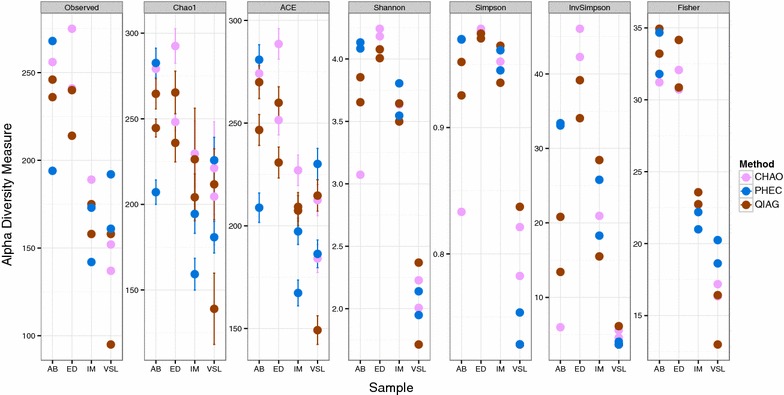
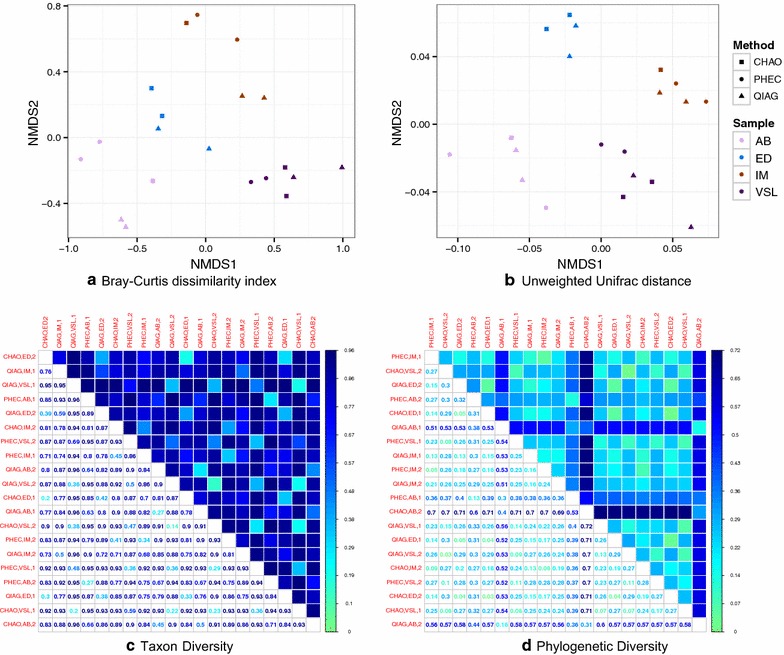
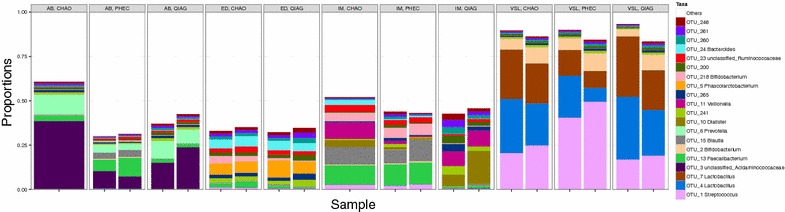
The CHAO yielded the highest and the QIAG kit the lowest amount of double-stranded DNA, but the purity of isolated nucleic acids was better for the latter method. The CHAO method yielded a higher concentration of bacterial taxa per mass (g) of faeces. Sequencing coverage was higher in CHAO method but a higher proportion of the initial sequencing reads were retained for assignments to operational taxonomic unit (OTU) in the QIAG kit compared to the other methods. The QIAG kit appeared to have longer trimmed reads and shorter regions of worse quality than the other two methods. A distinct separation of α-diversity indices between different DNA extraction methods was not observed. When compositional dissimilarities between samples were explored, a strong separation was observed according to sample type. The effect of the extraction method was either marginal (Bray-Curtis distance) or none (unweighted Unifrac distance). Taxon membership and abundance in each sample was independent of the DNA extraction method used.
We have benchmarked several DNA extraction methods commonly used in gut microbiota research and their differences depended on the downstream applications intended for use. Caution should be paid when the intention is to pool and analyse samples or data from studies which have used different DNA extraction methods.
传统和现代DNA提取方法对研究肠道微生物群在健康与疾病中作用的应用影响是当前备受关注的话题。使用三种常用方法从三个粪便样本和一个益生菌胶囊中提取基因组DNA;即离液剂(CHAO)法、苯酚/氯仿(PHEC)提取法、专用试剂盒(QIAG)。评估了这些方法在DNA产量和质量、使用定量PCR分析微生物群组成、16S rRNA基因深度测序以及测序分析流程方面的性能。
CHAO法产生的双链DNA产量最高,QIAG试剂盒产生的产量最低,但后一种方法分离出的核酸纯度更高。CHAO法每克粪便产生的细菌类群浓度更高。CHAO法的测序覆盖率更高,但与其他方法相比,QIAG试剂盒中保留用于分配到操作分类单元(OTU)的初始测序读数比例更高。与其他两种方法相比,QIAG试剂盒的修剪读数似乎更长,质量较差的区域更短。未观察到不同DNA提取方法之间α-多样性指数的明显分离。在探索样本之间的组成差异时,根据样本类型观察到明显的分离。提取方法的影响要么很小(Bray-Curtis距离),要么没有影响(非加权UniFrac距离)。每个样本中的分类单元成员和丰度与所使用的DNA提取方法无关。
我们对肠道微生物群研究中常用的几种DNA提取方法进行了基准测试,其差异取决于预期的下游应用。当打算汇总和分析来自使用不同DNA提取方法的研究的样本或数据时,应谨慎行事。