The Gut Health and Food Safety Programme, Quadram Institute Bioscience, Norwich Research Park, Colney, Norwich, UK.
Leeds Institute for Biomedical and Clinical Sciences, University of Leeds, Leeds, UK.
BMC Genomics. 2017 Nov 2;18(1):841. doi: 10.1186/s12864-017-4229-x.
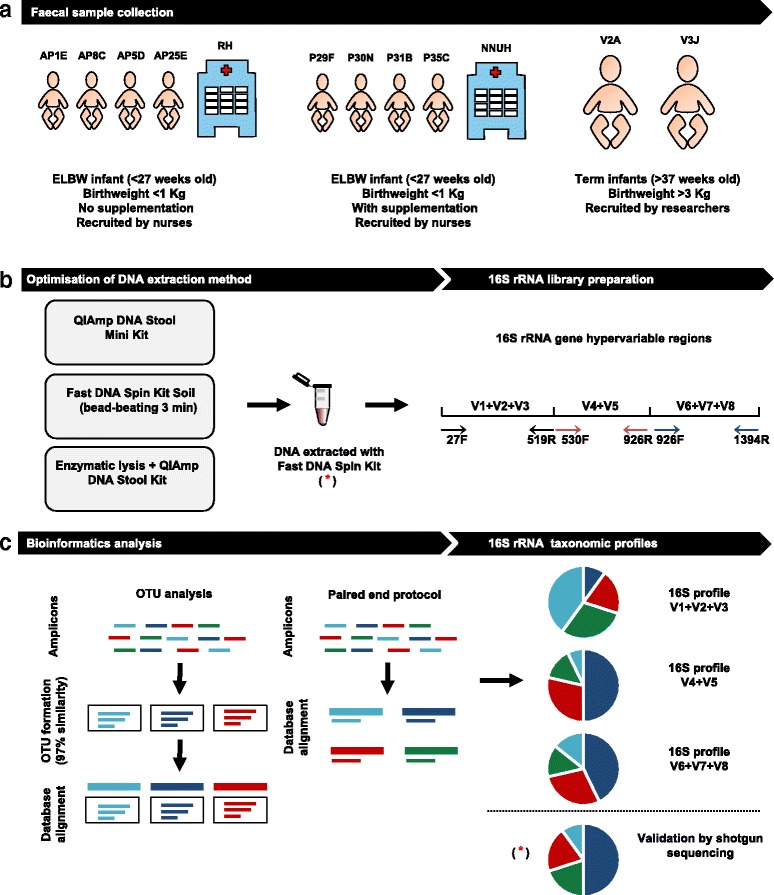
Infants born prematurely, particularly extremely low birth weight infants (ELBW) have altered gut microbial communities. Factors such as maternal health, gut immaturity, delivery mode, and antibiotic treatments are associated with microbiota disturbances, and are linked to an increased risk of certain diseases such as necrotising enterocolitis. Therefore, there is a requirement to optimally characterise microbial profiles in this at-risk cohort, via standardisation of methods, particularly for studying the influence of microbiota therapies (e.g. probiotic supplementation) on community profiles and health outcomes. Profiling of faecal samples using the 16S rRNA gene is a cost-efficient method for large-scale clinical studies to gain insights into the gut microbiota and additionally allows characterisation of cohorts were sample quantities are compromised (e.g. ELBW infants). However, DNA extraction method, and the 16S rRNA region targeted can significantly change bacterial community profiles obtained, and so confound comparisons between studies. Thus, we sought to optimise a 16S rRNA profiling protocol to allow standardisation for studying ELBW infant faecal samples, with or without probiotic supplementation.
Using ELBW faecal samples, we compared three different DNA extraction methods, and subsequently PCR amplified and sequenced three hypervariable regions of the 16S rRNA gene (V1 + V2 + V3), (V4 + V5) and (V6 + V7 + V8), and compared two bioinformatics approaches to analyse results (OTU and paired end). Paired shotgun metagenomics was used as a 'gold-standard'.
Results indicated a longer bead-beating step was required for optimal bacterial DNA extraction and that sequencing regions (V1 + V2 + V3) and (V6 + V7 + V8) provided the most representative taxonomic profiles, which was confirmed via shotgun analysis. Samples sequenced using the (V4 + V5) region were found to be underrepresented in specific taxa including Bifidobacterium, and had altered diversity profiles. Both bioinformatics 16S rRNA pipelines used in this study (OTU and paired end) presented similar taxonomic profiles at genus level.
We determined that DNA extraction from ELBW faecal samples, particularly those infants receiving probiotic supplementation, should include a prolonged beat-beating step. Furthermore, use of the 16S rRNA (V1 + V2 + V3) and (V6 + V7 + V8) regions provides reliable representation of ELBW microbiota profiles, while inclusion of the (V4 + V5) region may not be appropriate for studies where Bifidobacterium constitutes a resident microbiota member.
早产儿,尤其是极低出生体重儿(ELBW)的肠道微生物群落发生了改变。母亲健康状况、肠道不成熟、分娩方式和抗生素治疗等因素与微生物群紊乱有关,并与某些疾病(如坏死性小肠结肠炎)的风险增加有关。因此,需要通过方法标准化来优化该高危队列的微生物特征,特别是用于研究微生物组疗法(例如益生菌补充)对群落特征和健康结果的影响。使用 16S rRNA 基因对粪便样本进行分析是一种具有成本效益的方法,可用于大规模临床研究,以深入了解肠道微生物组,并且还可以对样本数量受到限制的队列进行特征描述(例如,ELBW 婴儿)。然而,DNA 提取方法和靶向的 16S rRNA 区域会显著改变获得的细菌群落特征,从而混淆了研究之间的比较。因此,我们试图优化 16S rRNA 分析方案,以允许对 ELBW 婴儿粪便样本进行标准化研究,无论是否添加益生菌。
使用 ELBW 粪便样本,我们比较了三种不同的 DNA 提取方法,随后对 16S rRNA 基因的三个高变区(V1+V2+V3)、(V4+V5)和(V6+V7+V8)进行了 PCR 扩增和测序,并比较了两种生物信息学方法来分析结果(OTU 和配对末端)。 shotgun 宏基因组学被用作“金标准”。
结果表明,需要更长的珠磨步骤才能获得最佳的细菌 DNA 提取效果,并且测序区域(V1+V2+V3)和(V6+V7+V8)提供了最具代表性的分类群特征,这通过 shotgun 分析得到了证实。使用(V4+V5)区域进行测序的样本在特定分类群(包括双歧杆菌)中被发现代表性不足,并且多样性特征发生了改变。本研究中使用的两种 16S rRNA 生物信息学分析管道(OTU 和配对末端)在属水平上呈现出相似的分类群特征。
我们确定,从 ELBW 粪便样本中提取 DNA,特别是那些接受益生菌补充的婴儿,应包括延长的珠磨步骤。此外,使用 16S rRNA(V1+V2+V3)和(V6+V7+V8)区域可可靠地代表 ELBW 微生物组特征,而包含(V4+V5)区域可能不适合研究双歧杆菌构成常驻微生物群成员的情况。