Department of Biology, University of Waterloo, Waterloo, Ontario, Canada.
BMC Bioinformatics. 2012 Feb 14;13:31. doi: 10.1186/1471-2105-13-31.
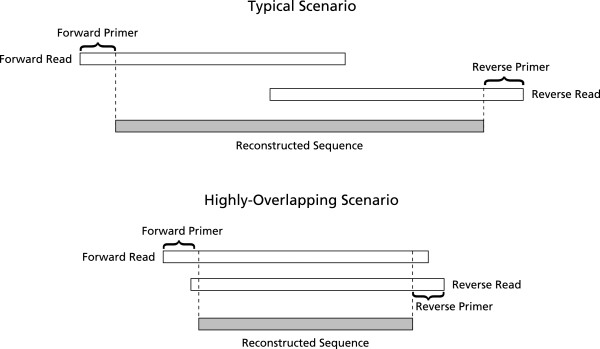
Illumina paired-end reads are used to analyse microbial communities by targeting amplicons of the 16S rRNA gene. Publicly available tools are needed to assemble overlapping paired-end reads while correcting mismatches and uncalled bases; many errors could be corrected to obtain higher sequence yields using quality information.
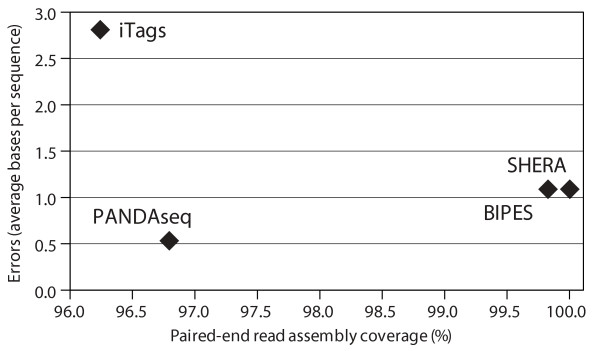
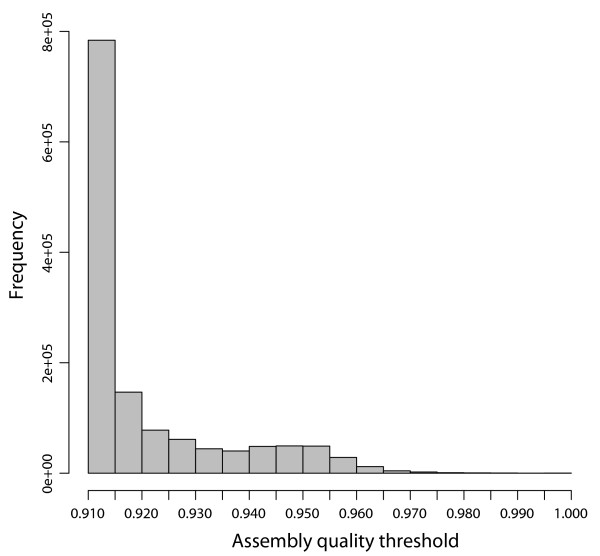
PANDAseq assembles paired-end reads rapidly and with the correction of most errors. Uncertain error corrections come from reads with many low-quality bases identified by upstream processing. Benchmarks were done using real error masks on simulated data, a pure source template, and a pooled template of genomic DNA from known organisms. PANDAseq assembled reads more rapidly and with reduced error incorporation compared to alternative methods.
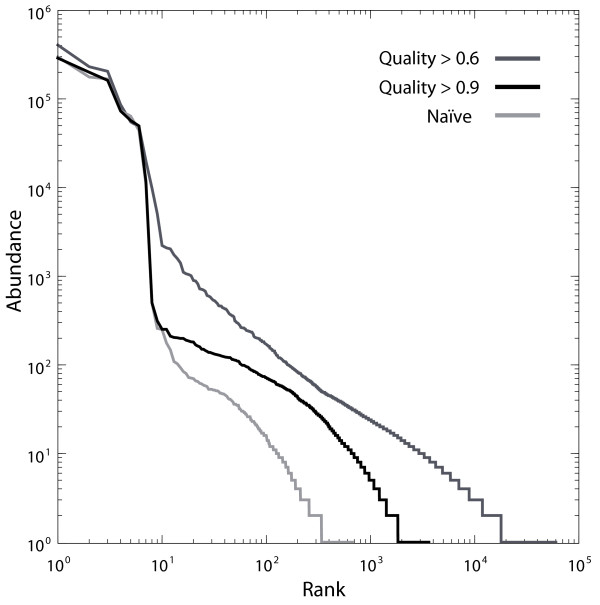
PANDAseq rapidly assembles sequences and scales to billions of paired-end reads. Assembly of control libraries showed a 4-50% increase in the number of assembled sequences over naïve assembly with negligible loss of "good" sequence.
Illumina 配对末端读取用于通过靶向 16S rRNA 基因的扩增子来分析微生物群落。需要公共可用的工具来组装重叠的配对末端读取,同时纠正错配和未呼叫的碱基;使用质量信息可以纠正许多错误以获得更高的序列产量。
PANDAseq 快速组装并纠正了大多数错误的配对末端读取。不确定的错误纠正来自上游处理识别的许多低质量碱基的读取。使用模拟数据、纯源模板和已知生物的基因组 DNA 混合模板上的真实错误掩模进行基准测试。与替代方法相比,PANDAseq 更快地组装了读取并减少了错误的掺入。
PANDAseq 快速组装序列并扩展到数十亿对末端读取。对照文库的组装显示,组装序列的数量比天真组装增加了 4-50%,而“良好”序列的损失可以忽略不计。