Saleh Suha, Lu Hao K, Evans Vanessa, Harisson David, Zhou Jingling, Jaworowski Anthony, Sallmann Georgina, Cheong Karey Y, Mota Talia M, Tennakoon Surekha, Angelovich Thomas A, Anderson Jenny, Harman Andrew, Cunningham Anthony, Gray Lachlan, Churchill Melissa, Mak Johnson, Drummer Heidi, Vatakis Dimitrios N, Lewin Sharon R, Cameron Paul U
Doherty Institute for Infection and Immunity, The University of Melbourne, Melbourne, Australia.
Department of Infectious Diseases, Alfred Health, Monash University, Melbourne, Australia.
Retrovirology. 2016 Jul 26;13(1):49. doi: 10.1186/s12977-016-0284-7.
Eradication of HIV cannot be achieved with combination antiretroviral therapy (cART) because of the persistence of long-lived latently infected resting memory CD4(+) T cells. We previously reported that HIV latency could be established in resting CD4(+) T cells in the presence of the chemokine CCL19. To define how CCL19 facilitated the establishment of latent HIV infection, the role of chemokine receptor signalling was explored.
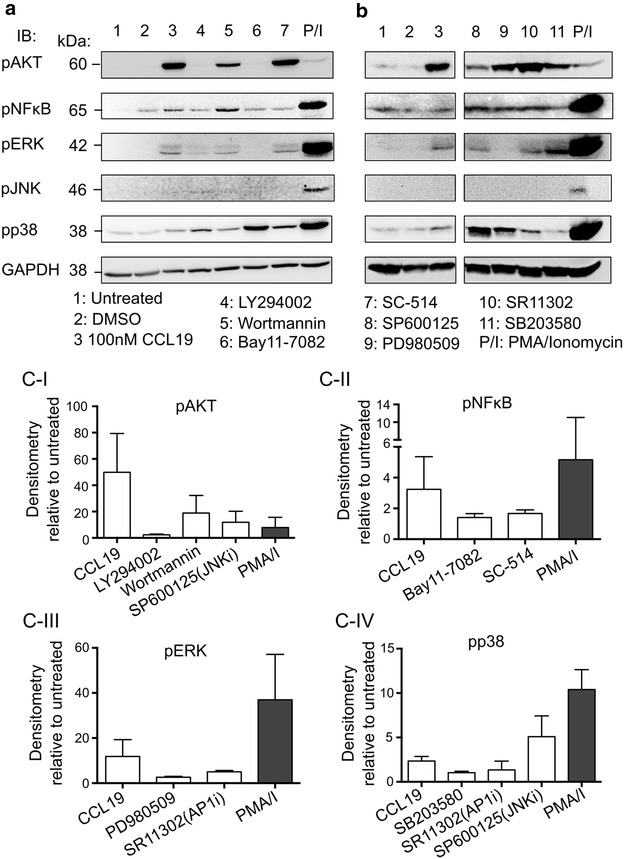
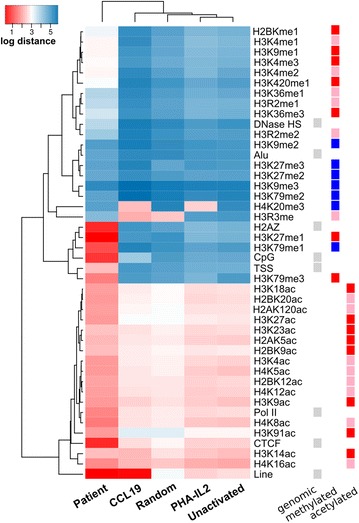
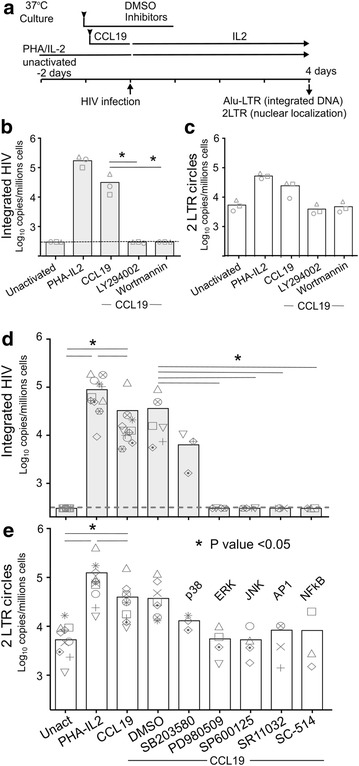
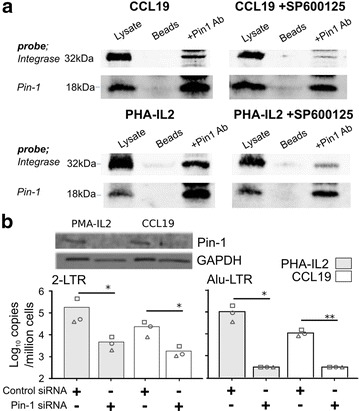
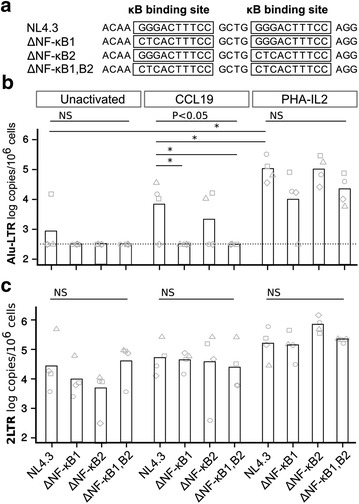
In resting CD4(+) T cells, CCL19 induced phosphorylation of RAC-alpha serine/threonine-protein kinase (Akt), nuclear factor kappa B (NF-κB), extracellular-signal-regulated kinase (ERK) and p38. Inhibition of the phosphoinositol-3-kinase (PI3K) and Ras/Raf/Mitogen-activated protein kinase/ERK kinase (MEK)/ERK signalling pathways inhibited HIV integration, without significant reduction in HIV nuclear entry (measured by Alu-LTR and 2-LTR circle qPCR respectively). Inhibiting activation of MEK1/ERK1/2, c-Jun N-terminal kinase (JNK), activating protein-1 (AP-1) and NF-κB, but not p38, also inhibited HIV integration. We also show that HIV integrases interact with Pin1 in CCL19-treated CD4(+) T cells and inhibition of JNK markedly reduced this interaction, suggesting that CCL19 treatment provided sufficient signals to protect HIV integrase from degradation via the proteasome pathway. Infection of CCL19-treated resting CD4(+) T cells with mutant strains of HIV, lacking NF-κB binding sites in the HIV long terminal repeat (LTR) compared to infection with wild type virus, led to a significant reduction in integration by up to 40-fold (range 1-115.4, p = 0.03). This was in contrast to only a modest reduction of 5-fold (range 1.7-11, p > 0.05) in fully activated CD4(+) T cells infected with the same mutants. Finally, we demonstrated significant differences in integration sites following HIV infection of unactivated, CCL19-treated, and fully activated CD4(+) T cells.
HIV integration in CCL19-treated resting CD4(+) T cells depends on NF-κB signalling and increases the stability of HIV integrase, which allow subsequent integration and establishment of latency. These findings have implications for strategies needed to prevent the establishment, and potentially reverse, latent infection.
由于长期潜伏感染的静息记忆CD4(+) T细胞持续存在,联合抗逆转录病毒疗法(cART)无法实现HIV的根除。我们之前报道过,在趋化因子CCL19存在的情况下,HIV潜伏期可在静息CD4(+) T细胞中建立。为了确定CCL19如何促进潜伏性HIV感染的建立,我们探讨了趋化因子受体信号传导的作用。
在静息CD4(+) T细胞中,CCL19诱导RAC-α丝氨酸/苏氨酸蛋白激酶(Akt)、核因子κB(NF-κB)、细胞外信号调节激酶(ERK)和p38磷酸化。磷酸肌醇-3-激酶(PI3K)和Ras/Raf/丝裂原活化蛋白激酶/ERK激酶(MEK)/ERK信号通路的抑制会抑制HIV整合,而HIV核进入(分别通过Alu-LTR和2-LTR环定量PCR测量)没有显著降低。抑制MEK1/ERK1/2、c-Jun氨基末端激酶(JNK)、活化蛋白-1(AP-1)和NF-κB的激活,但不抑制p38,也会抑制HIV整合。我们还表明,在CCL19处理的CD4(+) T细胞中,HIV整合酶与Pin1相互作用,JNK的抑制显著降低了这种相互作用,这表明CCL19处理提供了足够的信号来保护HIV整合酶不通过蛋白酶体途径降解。与野生型病毒感染相比,用HIV突变株感染CCL19处理的静息CD4(+) T细胞,HIV长末端重复序列(LTR)中缺乏NF-κB结合位点,导致整合显著降低,高达40倍(范围1-115.4,p = 0.03)。这与用相同突变株感染的完全活化CD4(+) T细胞中仅适度降低5倍(范围1.7-11,p>0.05)形成对比。最后,我们证明了未活化、CCL19处理和完全活化的CD4(+) T细胞感染HIV后的整合位点存在显著差异。
CCL19处理的静息CD4(+) T细胞中的HIV整合依赖于NF-κB信号传导,并增加了HIV整合酶的稳定性,这允许随后进行整合并建立潜伏期。这些发现对预防潜伏感染的建立以及可能逆转潜伏感染所需的策略具有重要意义。