King Paula, Pham Long K, Waltz Shannon, Sphar Dan, Yamamoto Robert T, Conrad Douglas, Taplitz Randy, Torriani Francesca, Forsyth R Allyn
FLIR Systems, Inc., La Jolla, California, United States of America.
Singlera Genomics, Inc., La Jolla, California, United States of America.
PLoS One. 2016 Aug 2;11(8):e0160124. doi: 10.1371/journal.pone.0160124. eCollection 2016.
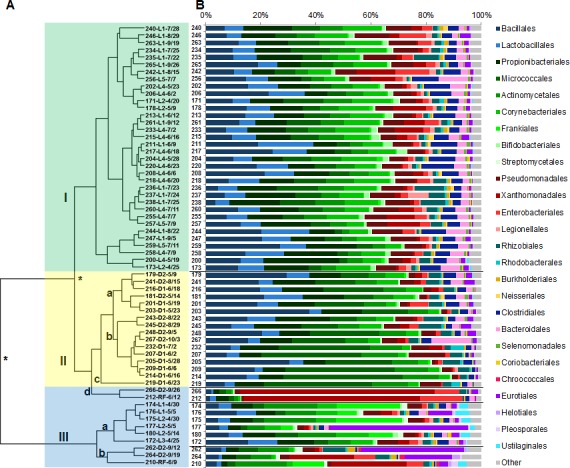
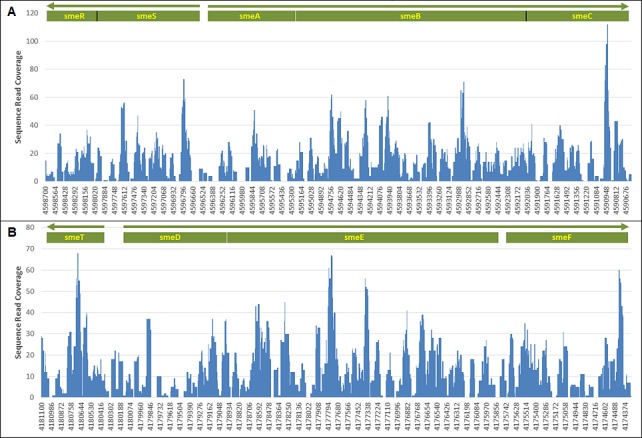
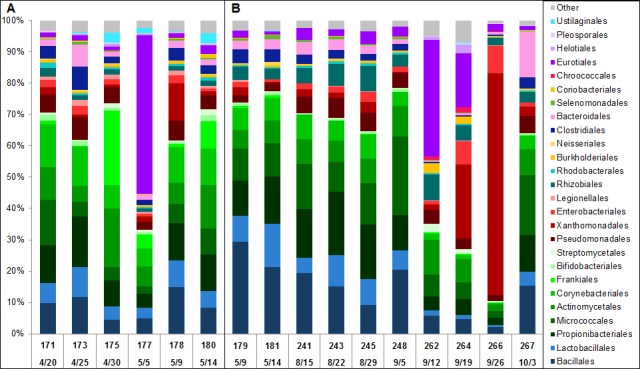
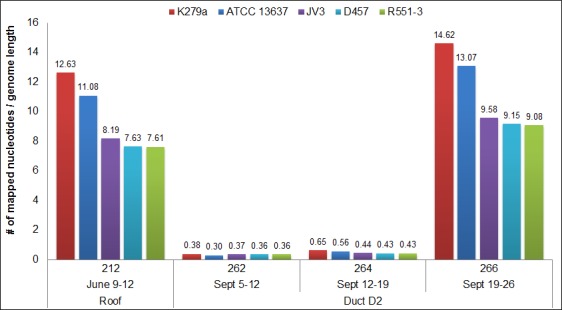
We describe the sampling of sixty-three uncultured hospital air samples collected over a six-month period and analysis using shotgun metagenomic sequencing. Our primary goals were to determine the longitudinal metagenomic variability of this environment, identify and characterize genomes of potential pathogens and determine whether they are atypical to the hospital airborne metagenome. Air samples were collected from eight locations which included patient wards, the main lobby and outside. The resulting DNA libraries produced 972 million sequences representing 51 gigabases. Hierarchical clustering of samples by the most abundant 50 microbial orders generated three major nodes which primarily clustered by type of location. Because the indoor locations were longitudinally consistent, episodic relative increases in microbial genomic signatures related to the opportunistic pathogens Aspergillus, Penicillium and Stenotrophomonas were identified as outliers at specific locations. Further analysis of microbial reads specific for Stenotrophomonas maltophilia indicated homology to a sequenced multi-drug resistant clinical strain and we observed broad sequence coverage of resistance genes. We demonstrate that a shotgun metagenomic sequencing approach can be used to characterize the resistance determinants of pathogen genomes that are uncharacteristic for an otherwise consistent hospital air microbial metagenomic profile.
我们描述了在六个月期间收集的63份未培养的医院空气样本的采样情况,并使用鸟枪法宏基因组测序进行分析。我们的主要目标是确定该环境的纵向宏基因组变异性,鉴定和表征潜在病原体的基因组,并确定它们是否与医院空气宏基因组不同。空气样本从包括病房、主大厅和室外在内的八个地点采集。产生的DNA文库产生了9.72亿条序列,代表510亿碱基。根据最丰富的50个微生物目对样本进行层次聚类,产生了三个主要节点,主要按地点类型聚类。由于室内地点在纵向上是一致的,与机会性病原体曲霉属、青霉属和嗜麦芽窄食单胞菌相关的微生物基因组特征的偶发性相对增加在特定地点被确定为异常值。对嗜麦芽窄食单胞菌特异的微生物读数的进一步分析表明与一株已测序的多重耐药临床菌株具有同源性,并且我们观察到耐药基因的广泛序列覆盖。我们证明,鸟枪法宏基因组测序方法可用于表征病原体基因组的耐药决定因素,这些因素对于原本一致的医院空气微生物宏基因组谱来说是不典型的。