Petereit Juli, Smith Sebastian, Harris Frederick C, Schlauch Karen A
University of Nevada, Reno, 1664 N. Virginia Street, Reno, 89557, USA.
BMC Syst Biol. 2016 Aug 1;10 Suppl 2(Suppl 2):51. doi: 10.1186/s12918-016-0298-8.
Networks provide effective models to study complex biological systems, such as gene and protein interaction networks. With the advent of new sequencing technologies, many life scientists are grasping for user-friendly methods and tools to examine biological components at the whole-systems level. Gene co-expression network analysis approaches are frequently used to successfully associate genes with biological processes and demonstrate great potential to gain further insights into the functionality of genes, thus becoming a standard approach in Systems Biology. Here the objective is to construct biologically meaningful and statistically strong co-expression networks, the identification of research dependent subnetworks, and the presentation of self-contained results.
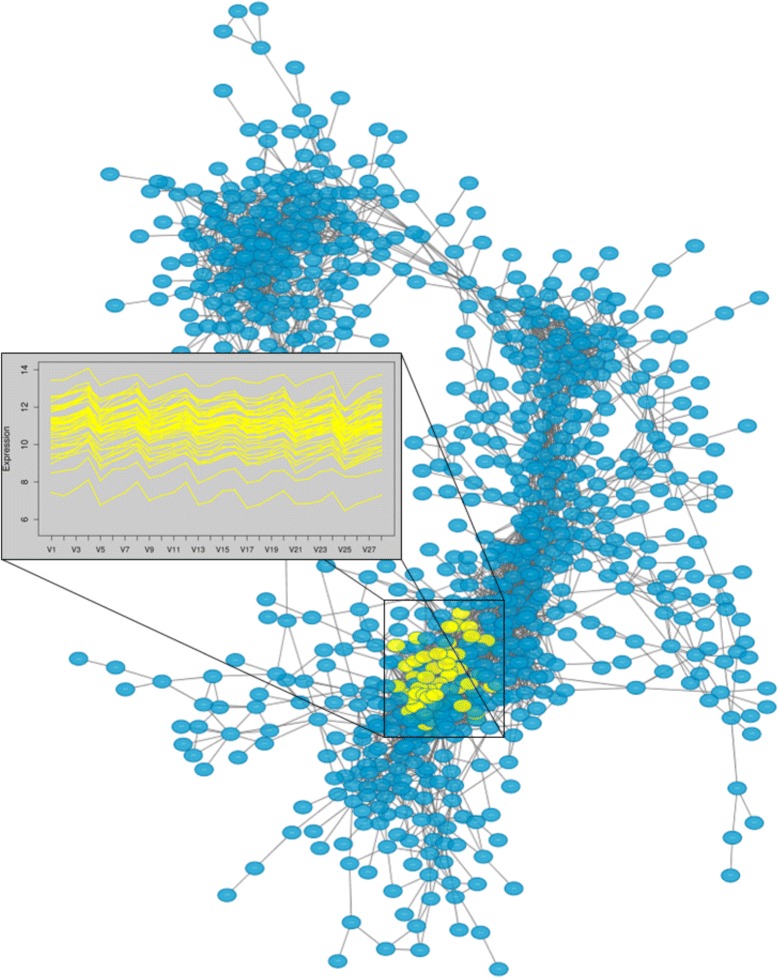
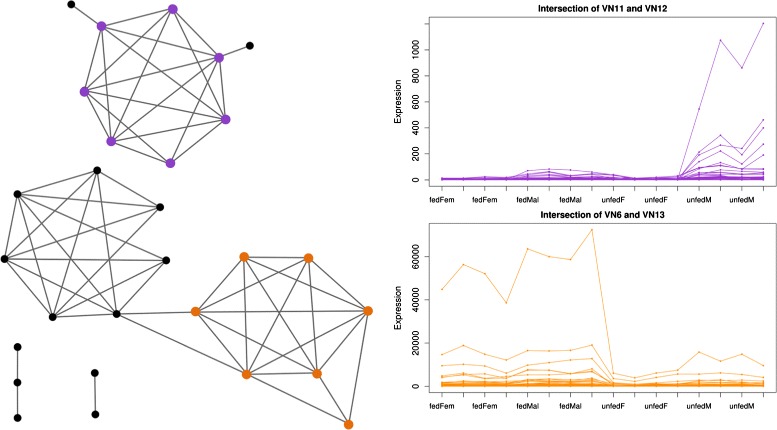
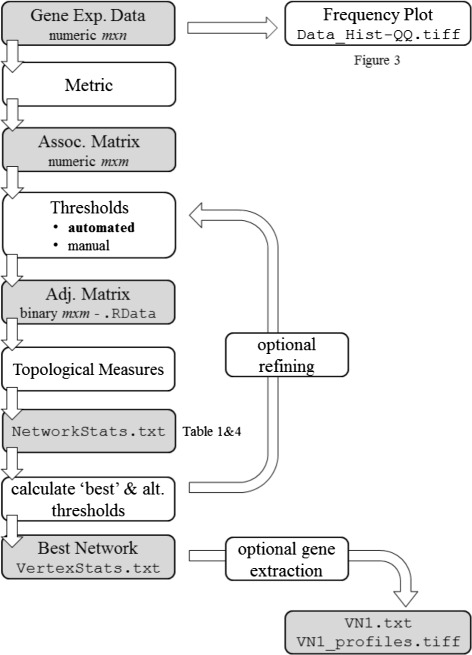
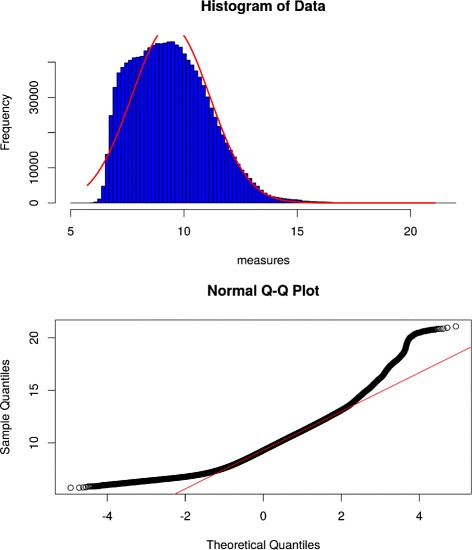
We introduce petal, a novel approach to generate gene co-expression network models based on experimental gene expression measures. petal focuses on statistical, mathematical, and biological characteristics of both, input data and output network models. Often over-looked issues of current co-expression analysis tools include the assumption of data normality, which is seldom the case for hight-throughput expression data obtained from RNA-seq technologies. petal does not assume data normality, making it a statistically appropriate method for RNA-seq data. Also, network models are rarely tested for their known typical architecture: scale-free and small-world. petal explicitly constructs networks based on both these characteristics, thereby generating biologically meaningful models. Furthermore, many network analysis tools require a number of user-defined input variables, these often require tuning and/or an understanding of the underlying algorithm; petal requires no user input other than experimental data. This allows for reproducible results, and simplifies the use of petal. Lastly, this approach is specifically designed for very large high-throughput datasets; this way, petal's network models represent as much of the entire system as possible to provide a whole-system approach.
petal is a novel tool for generating co-expression network models of whole-genomics experiments. It is implemented in R and available as a library. Its application to several whole-genome experiments has generated novel meaningful results and has lead the way to new testing hypothesizes for further biological investigation.
网络为研究复杂生物系统提供了有效的模型,如基因和蛋白质相互作用网络。随着新测序技术的出现,许多生命科学家正在寻求用户友好的方法和工具,以在全系统水平上研究生物成分。基因共表达网络分析方法经常被用于成功地将基因与生物过程联系起来,并显示出深入了解基因功能的巨大潜力,从而成为系统生物学中的一种标准方法。这里的目标是构建具有生物学意义和统计学强度的共表达网络,识别研究相关的子网,并呈现独立的结果。
我们介绍了petal,一种基于实验基因表达测量生成基因共表达网络模型的新方法。petal关注输入数据和输出网络模型的统计、数学和生物学特征。当前共表达分析工具经常被忽视的问题包括数据正态性假设,而从RNA测序技术获得的高通量表达数据很少符合这一假设。petal不假设数据正态性,使其成为适用于RNA测序数据的统计方法。此外,网络模型很少针对其已知的典型架构(无标度和小世界)进行测试。petal明确基于这两个特征构建网络,从而生成具有生物学意义的模型。此外,许多网络分析工具需要大量用户定义的输入变量,这些变量通常需要调整和/或理解底层算法;petal除了实验数据外不需要用户输入。这允许产生可重复的结果,并简化了petal的使用。最后,这种方法专门为非常大的高通量数据集设计;通过这种方式,petal的网络模型尽可能多地代表整个系统,以提供一种全系统方法。
petal是一种用于生成全基因组实验共表达网络模型的新工具。它在R语言中实现,并作为一个库可用。它在多个全基因组实验中的应用产生了新的有意义的结果,并为进一步的生物学研究开辟了新的测试假设之路。