Bend Eric G, Si Yue, Stevenson David A, Bayrak-Toydemir Pinar, Newcomb Tara M, Jorgensen Erik M, Swoboda Kathryn J
From the Department of Biology and Howard Hughes Medical Institute (E.G.B., E.M.J.), and Department of Pathology (Y.S., P.B.-T.), University of Utah, Salt Lake City; ARUP Institute for Clinical and Experimental Pathology (Y.S., P.B.-T.), Salt Lake City, UT; Division of Medical Genetics (D.A.S.), Department of Pediatrics, Stanford University, CA; Department of Neurology (T.M.N.), Pediatric Motor Disorders Research Program, University of Utah School of Medicine, Salt Lake City; and Department of Neurology (K.J.S.), Massachusetts General Hospital, Boston.
Neurology. 2016 Sep 13;87(11):1131-9. doi: 10.1212/WNL.0000000000003095. Epub 2016 Aug 24.
To perform genotype-phenotype analysis in an infant with congenital arthrogryposis due to a de novo missense mutation in the NALCN ion channel and explore the mechanism of pathogenicity using a Caenorhabditis elegans model.
We performed whole-exome sequencing in a preterm neonate with congenital arthrogryposis and a severe life-threatening clinical course. We examined the mechanism of pathogenicity of the associated NALCN mutation by engineering the orthologous mutation into the nematode C elegans using CRISPR-Cas9.
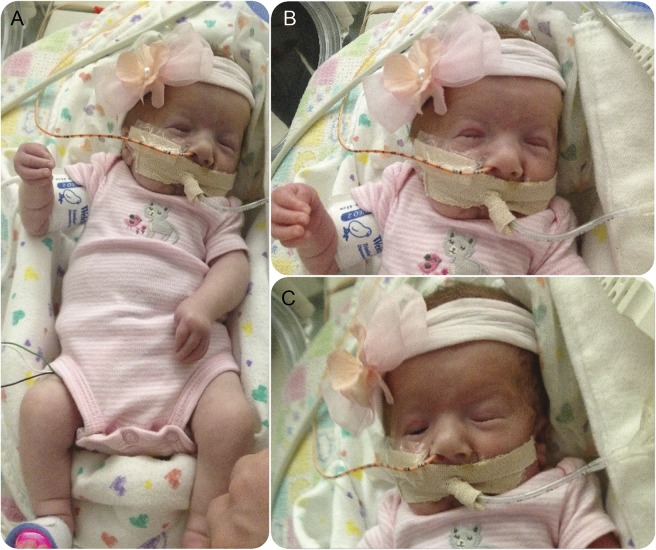
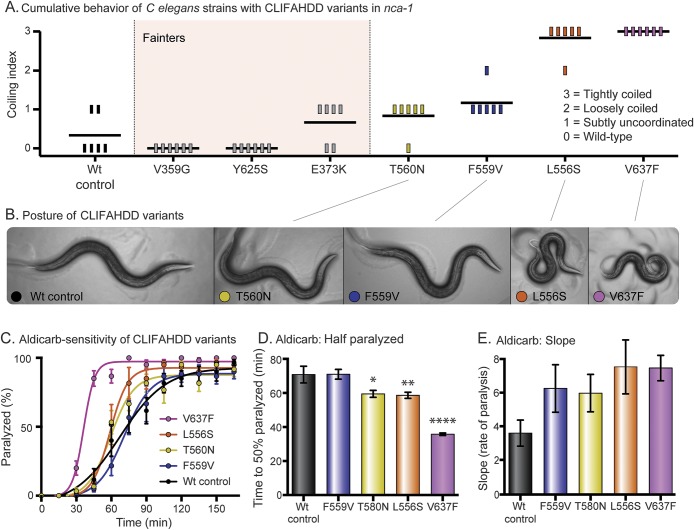
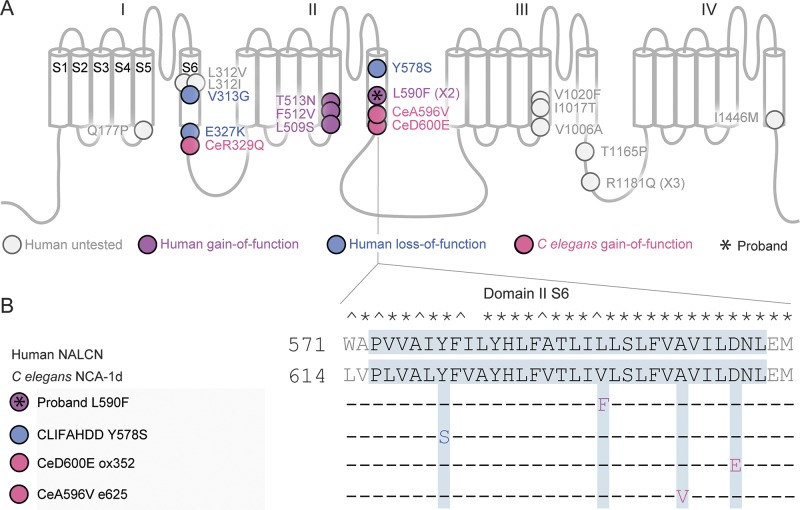
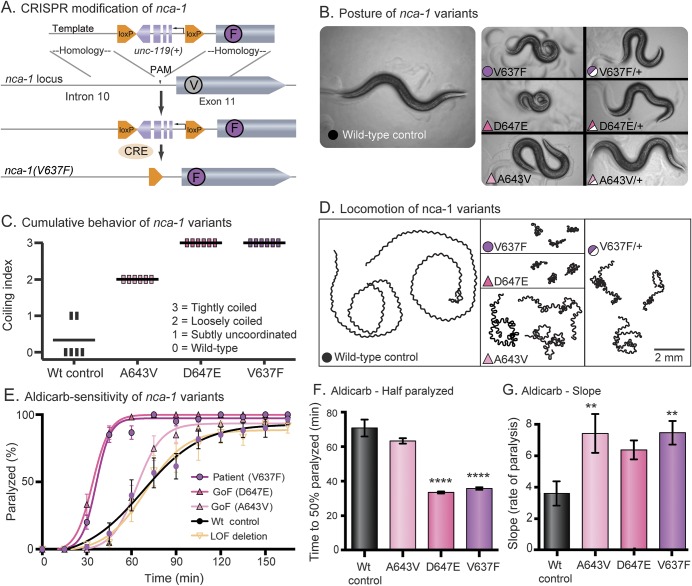
We identified a de novo missense mutation in NALCN, c.1768C>T, in an infant with a severe neonatal lethal form of the recently characterized CLIFAHDD syndrome (congenital contractures of the limbs and face with hypotonia and developmental delay). We report novel phenotypic features including prolonged episodes of stimulus-sensitive sustained muscular contraction associated with life-threatening episodes of desaturation and autonomic instability, extending the severity of previously described phenotypes associated with mutations in NALCN. When engineered into the C elegans ortholog, this mutation results in a severe gain-of-function phenotype, with hypercontraction and uncoordinated movement. We engineered 6 additional CLIFAHDD syndrome mutations into C elegans and the mechanism of action could be divided into 2 categories: half phenocopied gain-of-function mutants and half phenocopied loss-of-function mutants.
The clinical phenotype of our patient and electrophysiologic studies show sustained muscular contraction in response to transient sensory stimuli. In C elegans, this mutation causes neuronal hyperactivity via a gain-of-function NALCN ion channel. Testing human variants of NALCN in C elegans demonstrates that CLIFAHDD can be caused by dominant loss- or gain-of-function mutations in ion channel function.
对一名因NALCN离子通道新生错义突变导致先天性关节挛缩的婴儿进行基因型-表型分析,并利用秀丽隐杆线虫模型探索其致病机制。
我们对一名患有先天性关节挛缩且临床病程严重危及生命的早产儿进行了全外显子组测序。我们通过使用CRISPR-Cas9技术将直系同源突变引入线虫秀丽隐杆线虫中,研究相关NALCN突变的致病机制。
我们在一名患有近期发现的CLIFAHDD综合征(肢体和面部先天性挛缩伴肌张力减退和发育迟缓)严重新生儿致死型的婴儿中,鉴定出NALCN基因的一个新生错义突变,c.1768C>T。我们报告了新的表型特征,包括与危及生命的去饱和发作和自主神经不稳定相关的刺激敏感持续性肌肉收缩延长,扩展了先前描述的与NALCN突变相关表型的严重程度。当将该突变引入秀丽隐杆线虫直系同源基因时,会导致严重的功能获得性表型,出现过度收缩和运动不协调。我们将另外6个CLIFAHDD综合征突变引入秀丽隐杆线虫,其作用机制可分为两类:一半为功能获得性突变体的表型模拟,一半为功能丧失性突变体的表型模拟。
我们患者的临床表型和电生理研究表明,对短暂感觉刺激有持续的肌肉收缩反应。在秀丽隐杆线虫中,这种突变通过功能获得性NALCN离子通道导致神经元活动亢进。在秀丽隐杆线虫中测试NALCN的人类变体表明,CLIFAHDD可由离子通道功能的显性功能丧失或功能获得突变引起。