Kılıç Sefa, Erill Ivan
Department of Biological Sciences, University of Maryland Baltimore County (UMBC), Baltimore, MD, 21250, USA.
BMC Bioinformatics. 2016 Aug 31;17 Suppl 8(Suppl 8):277. doi: 10.1186/s12859-016-1113-7.
Comparative genomics can leverage the vast amount of available genomic sequences to reconstruct and analyze transcriptional regulatory networks in Bacteria, but the efficacy of this approach hinges on the ability to transfer regulatory network information from reference species to the genomes under analysis. Several methods have been proposed to transfer regulatory information between bacterial species, but the paucity and distributed nature of experimental information on bacterial transcriptional networks have prevented their systematic evaluation.
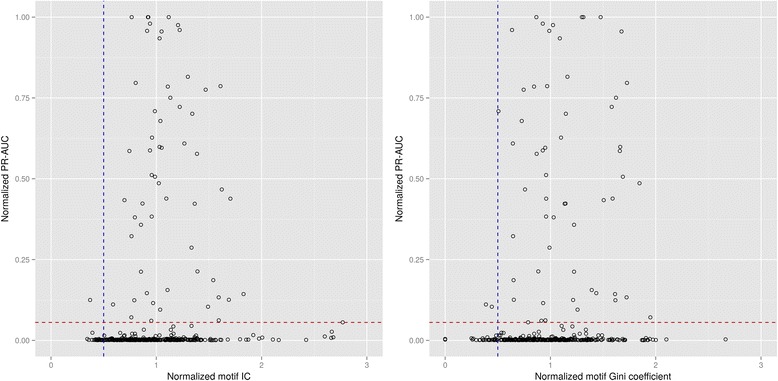
We report the compilation of a large catalog of transcription factor-binding sites across Bacteria and its use to systematically benchmark proposed transfer methods across pairs of bacterial species. We evaluate motif- and accuracy-based metrics to assess the results of regulatory network transfer and we identify the precision-recall area-under-the-curve as the best metric for this purpose due to the large class-imbalanced nature of the problem. Methods assuming conservation of the transcription factor-binding motif (motif-based) are shown to substantially outperform those assuming conservation of regulon composition (network-based), even though their efficiency can decrease sharply with increasing phylogenetic distance. Variations of the basic motif-based transfer method do not yield significant improvements in transfer accuracy. Our results indicate that detection of a large enough number of regulated orthologs is critical for network-based transfer methods, but that relaxing orthology requirements does not improve results. Using the transcriptional regulators LexA and Fur as case examples, we also show how DNA-binding domain sequence similarity can yield confounding results as an indicator of transfer efficiency for motif-based methods.
Counter to standard practice, our evaluation of metrics to assess the efficiency of methods for regulatory network information transfer reveals that the area under precision-recall (PR) curves is a more precise and informative metric than that of receiver-operating-characteristic (ROC) curves, confirming similar findings in other class-imbalanced settings. Our systematic assessment of transfer methods reveals that simple approaches to both motif- and network-based transfer of regulatory information provide equal or better results than more elaborate methods. We also show that there are not effective predictors of transfer efficacy, substantiating the long-standing practice of manual curation in comparative genomics analyses.
比较基因组学可以利用大量现有的基因组序列来重建和分析细菌中的转录调控网络,但这种方法的有效性取决于将调控网络信息从参考物种转移到被分析基因组的能力。已经提出了几种在细菌物种之间转移调控信息的方法,但关于细菌转录网络的实验信息匮乏且分布分散,阻碍了对它们的系统评估。
我们报告了一个涵盖细菌转录因子结合位点的大型目录的汇编,并利用它对成对细菌物种间提出的转移方法进行系统的基准测试。我们评估基于基序和准确性的指标来评估调控网络转移的结果,并且由于该问题存在大量类不平衡的性质,我们确定精确召回率曲线下面积是用于此目的的最佳指标。结果表明,尽管基于转录因子结合基序保守性的方法(基于基序的方法)的效率会随着系统发育距离的增加而急剧下降,但它们的表现明显优于基于操纵子组成保守性的方法(基于网络的方法)。基于基序的基本转移方法的变体在转移准确性方面没有显著提高。我们的结果表明,检测到足够数量的受调控直系同源物对于基于网络的转移方法至关重要,但放宽直系同源性要求并不会改善结果。以转录调节因子LexA和Fur为例,我们还展示了DNA结合结构域序列相似性作为基于基序方法转移效率的指标可能会产生混淆结果。
与标准做法相反,我们对评估调控网络信息转移方法效率的指标进行的评估表明,精确召回率(PR)曲线下面积是比接受者操作特征(ROC)曲线更精确且更具信息量的指标,这证实了在其他类不平衡情况下的类似发现。我们对转移方法的系统评估表明,简单的基于基序和基于网络的调控信息转移方法比更复杂的方法能提供相同或更好的结果。我们还表明,不存在有效的转移效果预测指标,这证实了比较基因组学分析中长期以来的手动编辑做法合理性。