Boareto Marcelo, Caticha Nestor
Institute of Physics, University of São Paulo, São Paulo, SP 05508-900, Brazil.
Microarrays (Basel). 2014 Dec 16;3(4):340-51. doi: 10.3390/microarrays3040340.
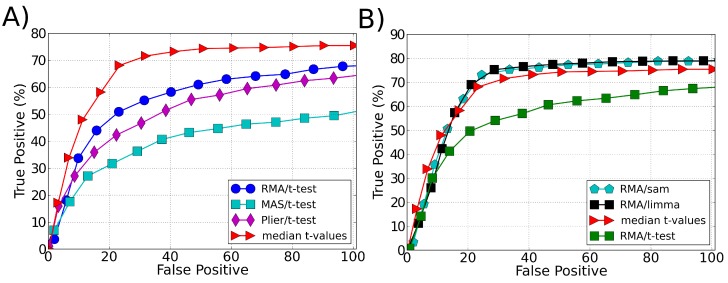
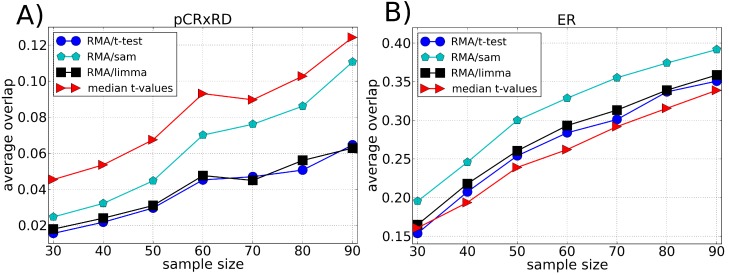
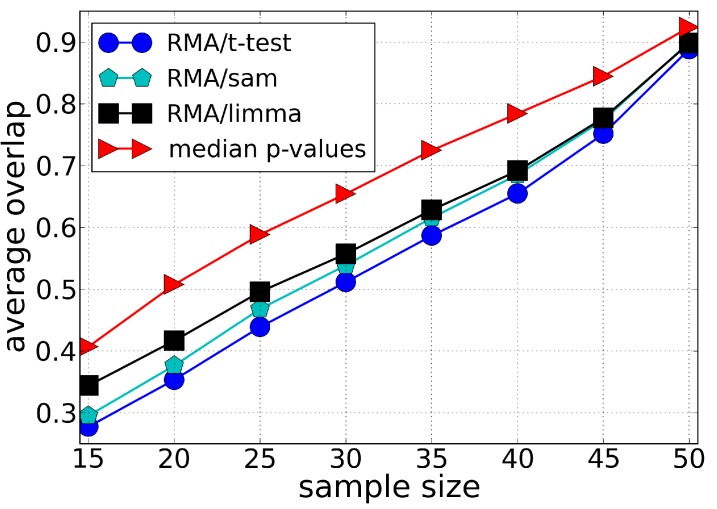
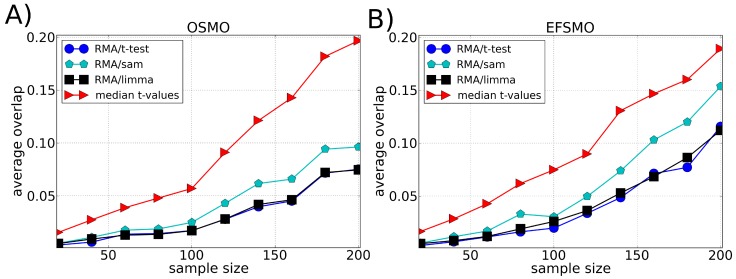
Microarray data analysis typically consists in identifying a list of differentially expressed genes (DEG), i.e., the genes that are differentially expressed between two experimental conditions. Variance shrinkage methods have been considered a better choice than the standard t-test for selecting the DEG because they correct the dependence of the error with the expression level. This dependence is mainly caused by errors in background correction, which more severely affects genes with low expression values. Here, we propose a new method for identifying the DEG that overcomes this issue and does not require background correction or variance shrinkage. Unlike current methods, our methodology is easy to understand and implement. It consists of applying the standard t-test directly on the normalized intensity data, which is possible because the probe intensity is proportional to the gene expression level and because the t-test is scale- and location-invariant. This methodology considerably improves the sensitivity and robustness of the list of DEG when compared with the t-test applied to preprocessed data and to the most widely used shrinkage methods, Significance Analysis of Microarrays (SAM) and Linear Models for Microarray Data (LIMMA). Our approach is useful especially when the genes of interest have small differences in expression and therefore get ignored by standard variance shrinkage methods.
微阵列数据分析通常在于识别一组差异表达基因(DEG),即那些在两种实验条件下差异表达的基因。对于选择DEG而言,方差缩减方法被认为比标准t检验是更好的选择,因为它们校正了误差与表达水平的相关性。这种相关性主要由背景校正中的误差引起,而背景校正误差对低表达值的基因影响更为严重。在此,我们提出一种识别DEG的新方法,该方法克服了这一问题,且不需要背景校正或方差缩减。与当前方法不同,我们的方法易于理解和实施。它包括直接对归一化强度数据应用标准t检验,这是可行的,因为探针强度与基因表达水平成正比,并且t检验是尺度不变和位置不变的。与应用于预处理数据的t检验以及最广泛使用的缩减方法微阵列显著性分析(SAM)和微阵列数据线性模型(LIMMA)相比,该方法显著提高了DEG列表的灵敏度和稳健性。我们的方法特别有用,尤其是当感兴趣的基因在表达上差异较小,因此会被标准方差缩减方法忽略时。