Hoenen Claire, Gustin Audrey, Birck Cindy, Kirchmeyer Mélanie, Beaume Nicolas, Felten Paul, Grandbarbe Luc, Heuschling Paul, Heurtaux Tony
Life Sciences Research Unit, Laboratory of Neurobiology, University of Luxembourg, Faculty of Science, Technology and Communication, 7, avenue des Hauts Fourneaux, L-4362, Esch-sur-Alzette, Luxembourg.
PLoS One. 2016 Sep 13;11(9):e0162717. doi: 10.1371/journal.pone.0162717. eCollection 2016.
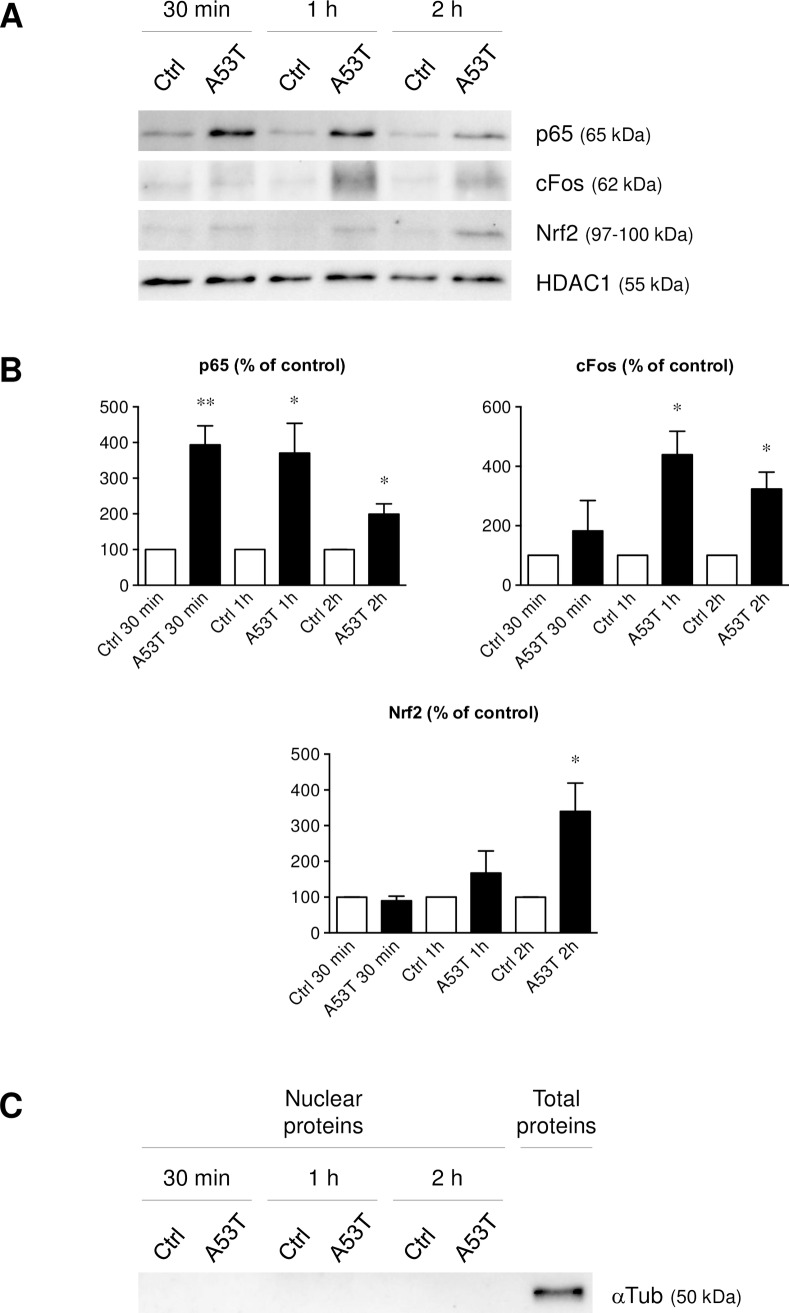
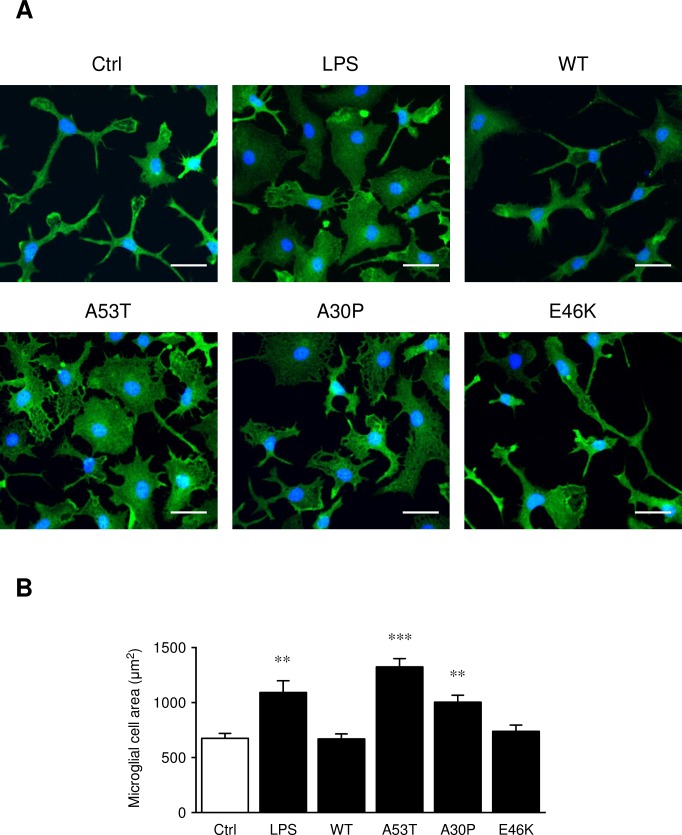
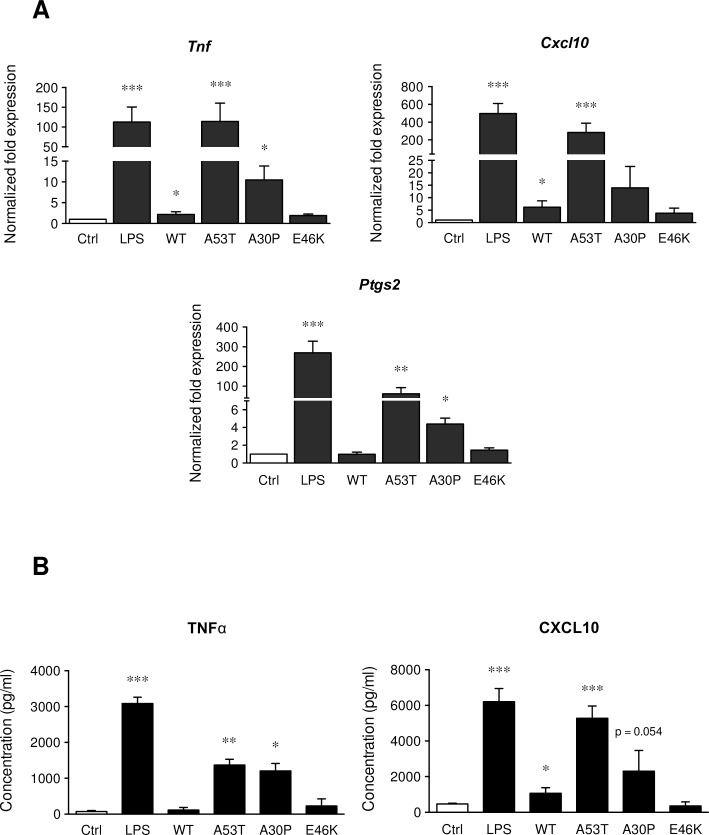
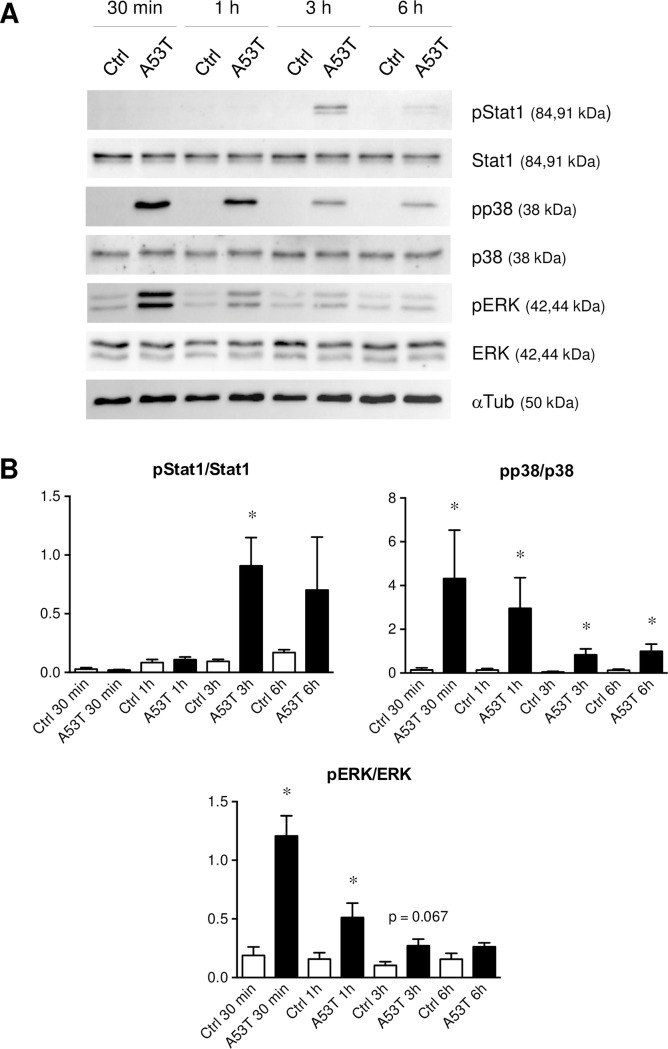
Parkinson's disease (PD) is histologically described by the deposition of α-synuclein, whose accumulation in Lewy bodies causes dopaminergic neuronal death. Although most of PD cases are sporadic, point mutations of the gene encoding the α-synuclein protein cause inherited forms of PD. There are currently six known point mutations that result in familial PD. Oxidative stress and neuroinflammation have also been described as early events associated with dopaminergic neuronal degeneration in PD. Though it is known that microglia are activated by wild-type α-synuclein, little is known about its mutated forms and the signaling cascades responsible for this microglial activation. The present study was designed to investigate consequences of wild-type and mutant α-synuclein (A53T, A30P and E46K) exposure on microglial reactivity. Interestingly, we described that α-synuclein-induced microglial reactivity appeared to be peptide-dependent. Indeed, the A53T protein activated more strongly microglia than the wild-type α-synuclein and other mutants. This A53T-induced microglial reactivity mechanism was found to depend on phosphorylation mechanisms mediated by MAPKs and on successive NFkB/AP-1/Nrf2 pathways activation. These results suggest that the microgliosis intensity during PD might depend on the type of α-synuclein protein implicated. Indeed, mutated forms are more potent microglial stimulators than wild-type α-synuclein. Based on these data, anti-inflammatory and antioxidant therapeutic strategies may be valid in order to reduce microgliosis but also to subsequently slow down PD progression, especially in familial cases.
帕金森病(PD)在组织学上表现为α-突触核蛋白的沉积,其在路易小体中的积累导致多巴胺能神经元死亡。虽然大多数PD病例是散发性的,但编码α-突触核蛋白的基因突变会导致遗传性PD。目前已知有六种点突变会导致家族性PD。氧化应激和神经炎症也被描述为与PD中多巴胺能神经元变性相关的早期事件。虽然已知小胶质细胞会被野生型α-突触核蛋白激活,但对其突变形式以及负责这种小胶质细胞激活的信号级联反应了解甚少。本研究旨在调查野生型和突变型α-突触核蛋白(A53T、A30P和E46K)暴露对小胶质细胞反应性的影响。有趣的是,我们发现α-突触核蛋白诱导的小胶质细胞反应性似乎是肽依赖性的。事实上,A53T蛋白比野生型α-突触核蛋白和其他突变体更强烈地激活小胶质细胞。发现这种A53T诱导的小胶质细胞反应性机制依赖于丝裂原活化蛋白激酶介导的磷酸化机制以及随后的NFkB/AP-1/Nrf2途径激活。这些结果表明,PD期间的小胶质细胞增生强度可能取决于所涉及的α-突触核蛋白的类型。事实上,突变形式比野生型α-突触核蛋白更有效地刺激小胶质细胞。基于这些数据,抗炎和抗氧化治疗策略可能是有效的,以减少小胶质细胞增生,进而减缓PD的进展,特别是在家族性病例中。