Meester Josephina A N, Vandeweyer Geert, Pintelon Isabel, Lammens Martin, Van Hoorick Lana, De Belder Simon, Waitzman Kathryn, Young Luciana, Markham Larry W, Vogt Julie, Richer Julie, Beauchesne Luc M, Unger Sheila, Superti-Furga Andrea, Prsa Milan, Dhillon Rami, Reyniers Edwin, Dietz Harry C, Wuyts Wim, Mortier Geert, Verstraeten Aline, Van Laer Lut, Loeys Bart L
Center of Medical Genetics, University of Antwerp and Antwerp University Hospital, Antwerp, Belgium.
Department of Cell Biology and Histology, University of Antwerp, Antwerp, Belgium.
Genet Med. 2017 Apr;19(4):386-395. doi: 10.1038/gim.2016.126. Epub 2016 Sep 15.
Thoracic aortic aneurysm and dissection (TAAD) is typically inherited in an autosomal dominant manner, but rare X-linked families have been described. So far, the only known X-linked gene is FLNA, which is associated with the periventricular nodular heterotopia type of Ehlers-Danlos syndrome. However, mutations in this gene explain only a small number of X-linked TAAD families.
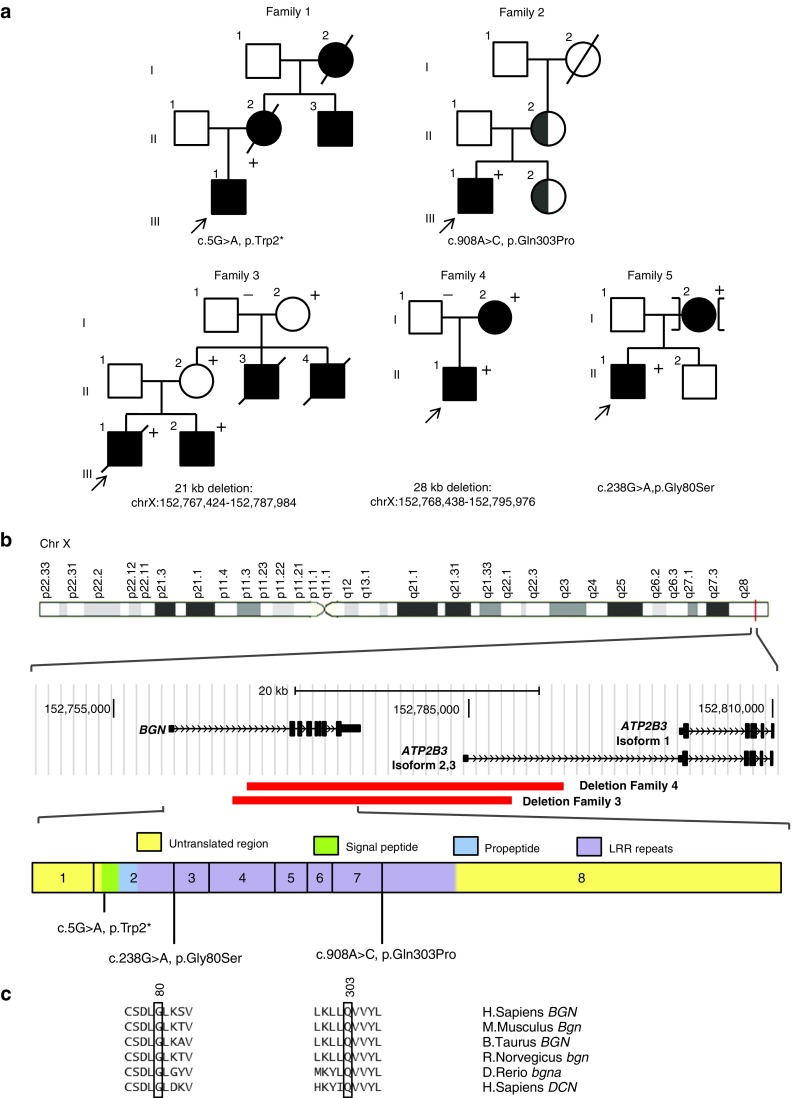
We performed targeted resequencing of 368 candidate genes in a cohort of 11 molecularly unexplained Marfan probands. Subsequently, Sanger sequencing of BGN in 360 male and 155 female molecularly unexplained TAAD probands was performed.
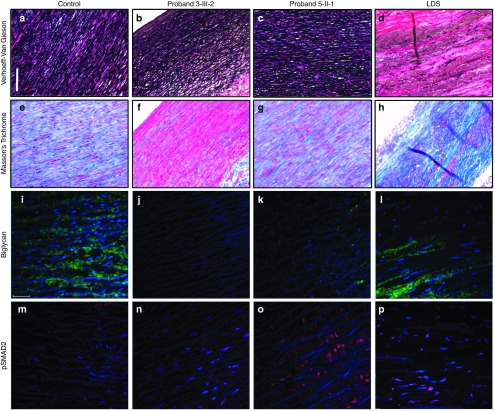
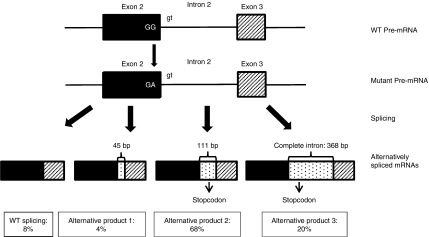
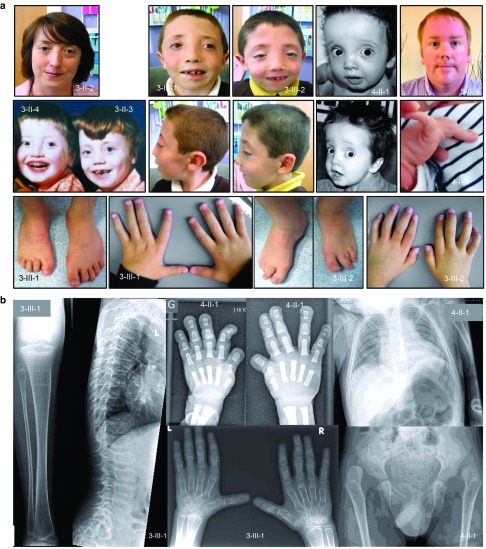
We found five individuals with loss-of-function mutations in BGN encoding the small leucine-rich proteoglycan biglycan. The clinical phenotype is characterized by early-onset aortic aneurysm and dissection. Other recurrent findings include hypertelorism, pectus deformity, joint hypermobility, contractures, and mild skeletal dysplasia. Fluorescent staining revealed an increase in TGF-β signaling, evidenced by an increase in nuclear pSMAD2 in the aortic wall. Our results are in line with those of prior reports demonstrating that Bgn-deficient male BALB/cA mice die from aortic rupture.
In conclusion, BGN gene defects in humans cause an X-linked syndromic form of severe TAAD that is associated with preservation of elastic fibers and increased TGF-β signaling.Genet Med 19 4, 386-395.
胸主动脉瘤和夹层(TAAD)通常以常染色体显性方式遗传,但也有罕见的X连锁家族被报道。到目前为止,唯一已知的X连锁基因是FLNA,它与埃勒斯-当洛综合征的室周结节性异位症有关。然而,该基因的突变仅能解释少数X连锁TAAD家族的病因。
我们对11例分子病因不明的马凡综合征先证者队列中的368个候选基因进行了靶向重测序。随后,对360例男性和155例女性分子病因不明的TAAD先证者进行了BGN基因的桑格测序。
我们发现5例个体在编码富含亮氨酸小分子蛋白聚糖双糖链蛋白聚糖的BGN基因中存在功能缺失突变。临床表型的特征为早发性主动脉瘤和夹层。其他常见表现包括眼距增宽、胸壁畸形、关节活动过度、挛缩和轻度骨骼发育异常。荧光染色显示主动脉壁中核pSMAD2增加,提示转化生长因子-β(TGF-β)信号传导增强。我们的结果与先前报道一致,即Bgn基因缺陷的雄性BALB/cA小鼠死于主动脉破裂。
总之,人类BGN基因缺陷会导致一种X连锁综合征形式的严重TAAD,其与弹性纤维的保留和TGF-β信号传导增强有关。《遗传医学》19 4, 386 - 395。