Li Mingzhou, Chen Lei, Tian Shilin, Lin Yu, Tang Qianzi, Zhou Xuming, Li Diyan, Yeung Carol K L, Che Tiandong, Jin Long, Fu Yuhua, Ma Jideng, Wang Xun, Jiang Anan, Lan Jing, Pan Qi, Liu Yingkai, Luo Zonggang, Guo Zongyi, Liu Haifeng, Zhu Li, Shuai Surong, Tang Guoqing, Zhao Jiugang, Jiang Yanzhi, Bai Lin, Zhang Shunhua, Mai Miaomiao, Li Changchun, Wang Dawei, Gu Yiren, Wang Guosong, Lu Hongfeng, Li Yan, Zhu Haihao, Li Zongwen, Li Ming, Gladyshev Vadim N, Jiang Zhi, Zhao Shuhong, Wang Jinyong, Li Ruiqiang, Li Xuewei
Institute of Animal Genetics and Breeding, College of Animal Science and Technology, Sichuan Agricultural University, Chengdu 611130, China.
Key Laboratory of Pig Industry Sciences (Ministry of Agriculture), Chongqing Academy of Animal Sciences, Chongqing 402460, China.
Genome Res. 2017 May;27(5):865-874. doi: 10.1101/gr.207456.116. Epub 2016 Sep 19.
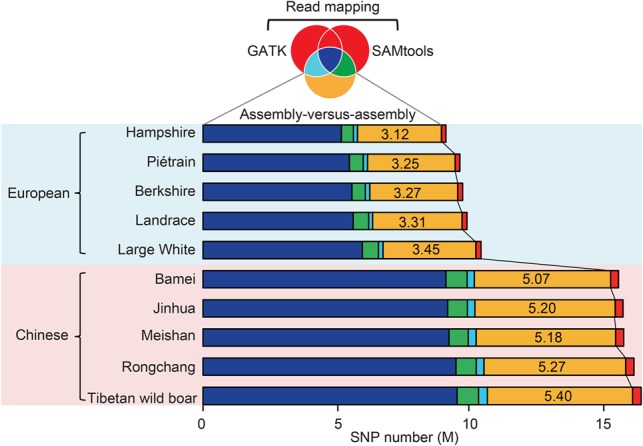
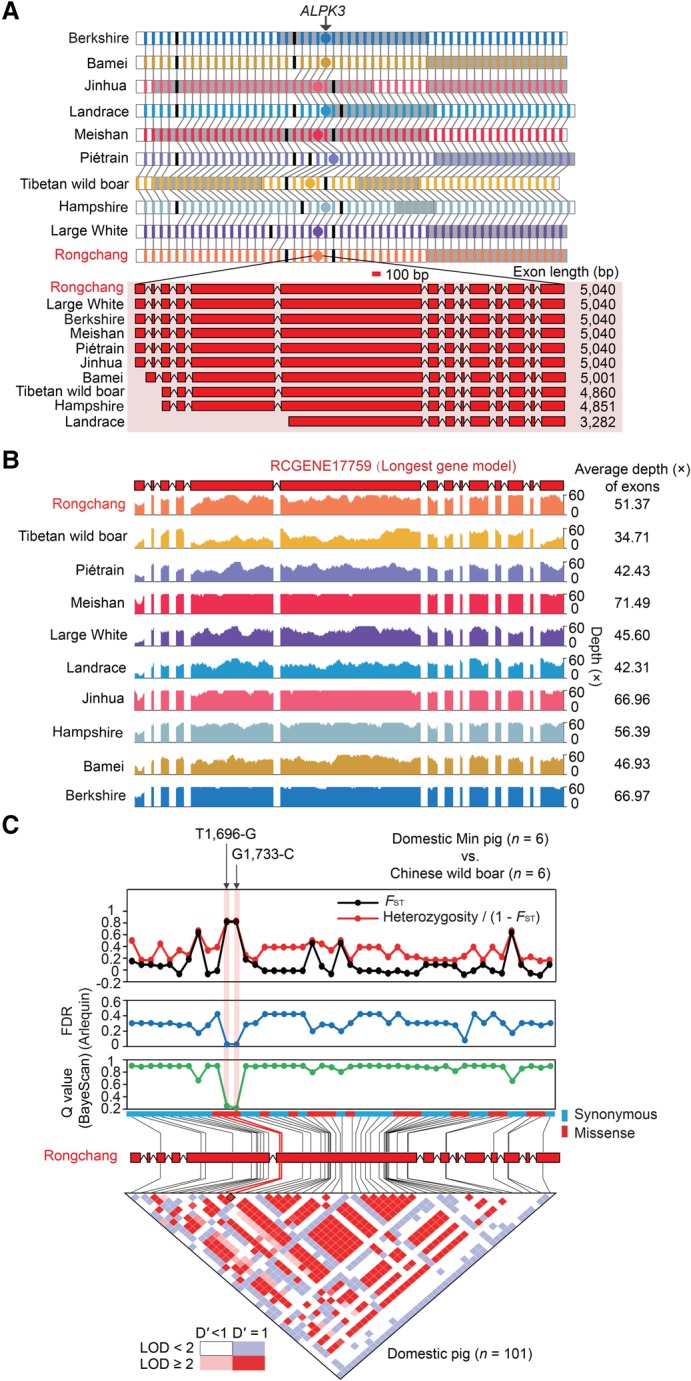
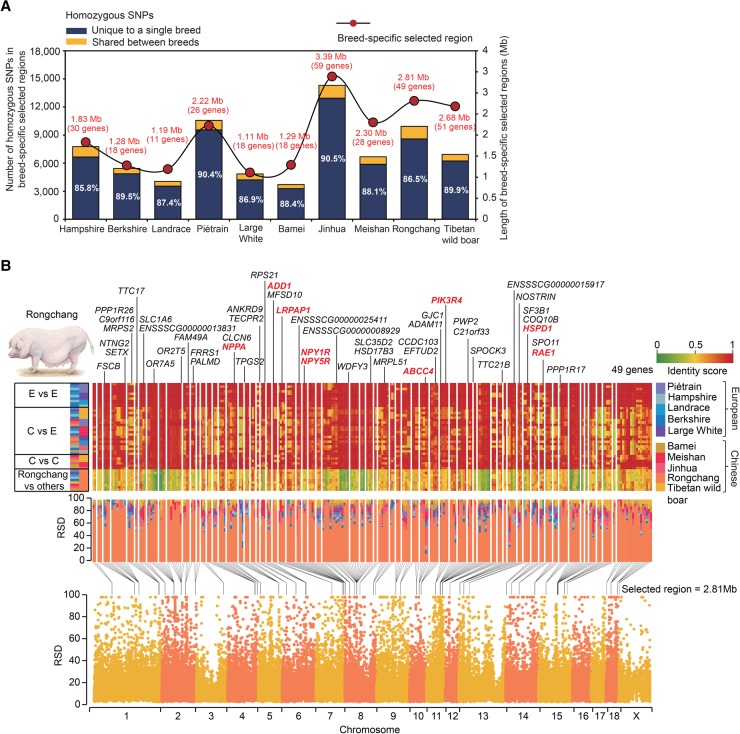
Uncovering genetic variation through resequencing is limited by the fact that only sequences with similarity to the reference genome are examined. Reference genomes are often incomplete and cannot represent the full range of genetic diversity as a result of geographical divergence and independent demographic events. To more comprehensively characterize genetic variation of pigs (), we generated de novo assemblies of nine geographically and phenotypically representative pigs from Eurasia. By comparing them to the reference pig assembly, we uncovered a substantial number of novel SNPs and structural variants, as well as 137.02-Mb sequences harboring 1737 protein-coding genes that were absent in the reference assembly, revealing variants left by selection. Our results illustrate the power of whole-genome de novo sequencing relative to resequencing and provide valuable genetic resources that enable effective use of pigs in both agricultural production and biomedical research.
通过重测序来揭示遗传变异受到限制,因为仅检测与参考基因组具有相似性的序列。参考基因组往往不完整,由于地理分化和独立的种群统计学事件,无法代表遗传多样性的全貌。为了更全面地表征猪的遗传变异,我们对来自欧亚大陆的9头具有地理和表型代表性的猪进行了从头组装。通过将它们与参考猪基因组组装进行比较,我们发现了大量新的单核苷酸多态性(SNP)和结构变异,以及137.02兆碱基的序列,其中包含1737个参考组装中不存在的蛋白质编码基因,揭示了选择留下的变异。我们的结果说明了全基因组从头测序相对于重测序的优势,并提供了宝贵的遗传资源,有助于在农业生产和生物医学研究中有效利用猪。