Barić Ivo, Staufner Christian, Augoustides-Savvopoulou Persephone, Chien Yin-Hsiu, Dobbelaere Dries, Grünert Sarah C, Opladen Thomas, Petković Ramadža Danijela, Rakić Bojana, Wedell Anna, Blom Henk J
Department of Pediatrics, University Hospital Center Zagreb, Kišpatićeva 12, Rebro, 10000, Zagreb, Croatia.
University of Zagreb, School of Medicine, Zagreb, Croatia.
J Inherit Metab Dis. 2017 Jan;40(1):5-20. doi: 10.1007/s10545-016-9972-7. Epub 2016 Sep 26.
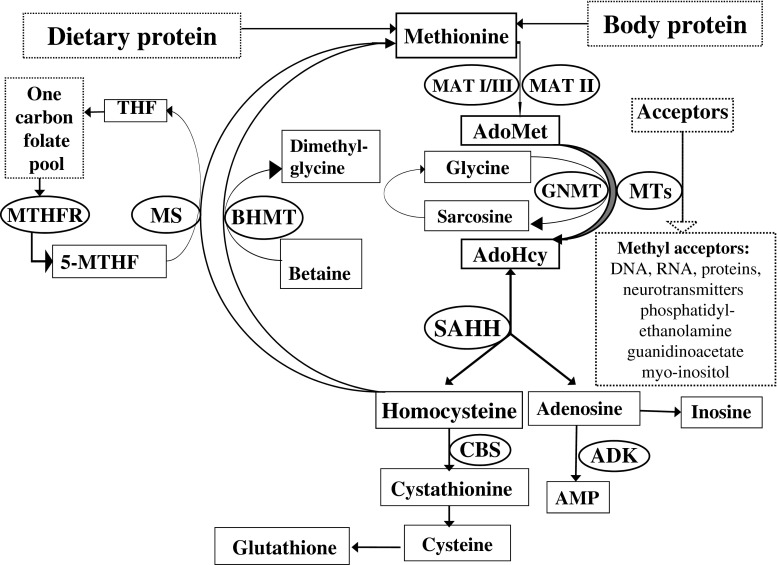
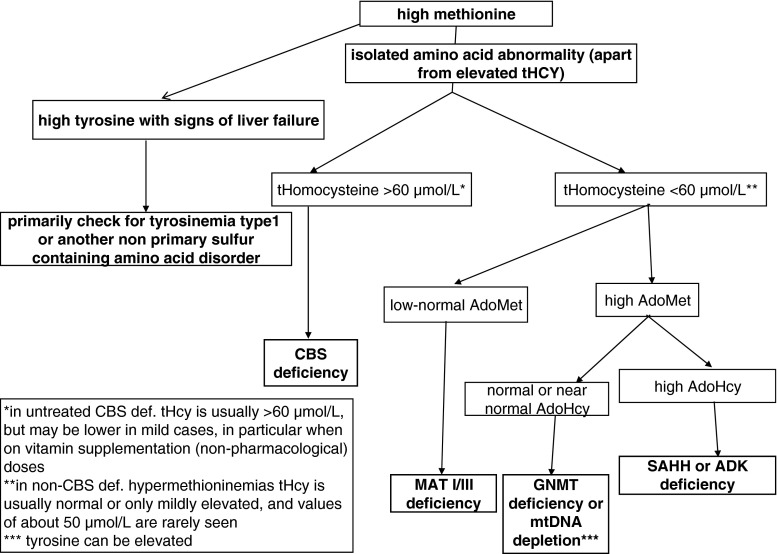
Inherited methylation disorders are a group of rarely reported, probably largely underdiagnosed disorders affecting transmethylation processes in the metabolic pathway between methionine and homocysteine. These are methionine adenosyltransferase I/III, glycine N-methyltransferase, S-adenosylhomocysteine hydrolase and adenosine kinase deficiencies. This paper provides the first consensus recommendations for the diagnosis and management of methylation disorders. Following search of the literature and evaluation according to the SIGN-methodology of all reported patients with methylation defects, graded recommendations are provided in a structured way comprising diagnosis (clinical presentation, biochemical abnormalities, differential diagnosis, newborn screening, prenatal diagnosis), therapy and follow-up. Methylation disorders predominantly affect the liver, central nervous system and muscles, but clinical presentation can vary considerably between and within disorders. Although isolated hypermethioninemia is the biochemical hallmark of this group of disorders, it is not always present, especially in early infancy. Plasma S-adenosylmethionine and S-adenosylhomocysteine are key metabolites for the biochemical clarification of isolated hypermethioninemia. Mild hyperhomocysteinemia can be present in all methylation disorders. Methylation disorders do not qualify as primary targets of newborn screening. A low-methionine diet can be beneficial in patients with methionine adenosyltransferase I/III deficiency if plasma methionine concentrations exceed 800 μmol/L. There is some evidence that this diet may also be beneficial in patients with S-adenosylhomocysteine hydrolase and adenosine kinase deficiencies. S-adenosylmethionine supplementation may be useful in patients with methionine adenosyltransferase I/III deficiency. Recommendations given in this article are based on general principles and in practice should be adjusted individually according to patient's age, severity of the disease, clinical and laboratory findings.
遗传性甲基化障碍是一组报道较少、可能在很大程度上未被诊断的疾病,影响蛋氨酸和同型半胱氨酸代谢途径中的转甲基过程。这些疾病包括蛋氨酸腺苷转移酶I/III、甘氨酸N-甲基转移酶、S-腺苷同型半胱氨酸水解酶和腺苷激酶缺乏症。本文首次提出了甲基化障碍诊断和管理的共识性建议。在检索文献并根据SIGN方法对所有报道的甲基化缺陷患者进行评估后,以结构化方式提供了分级建议,包括诊断(临床表现、生化异常、鉴别诊断、新生儿筛查、产前诊断)、治疗和随访。甲基化障碍主要影响肝脏、中枢神经系统和肌肉,但不同疾病之间以及同一疾病内部的临床表现可能有很大差异。虽然孤立性高蛋氨酸血症是这组疾病的生化标志,但并非总是存在,尤其是在婴儿早期。血浆S-腺苷蛋氨酸和S-腺苷同型半胱氨酸是孤立性高蛋氨酸血症生化诊断的关键代谢物。所有甲基化障碍患者都可能出现轻度高同型半胱氨酸血症。甲基化障碍不符合新生儿筛查的主要目标。如果血浆蛋氨酸浓度超过800μmol/L,低蛋氨酸饮食对蛋氨酸腺苷转移酶I/III缺乏症患者可能有益。有证据表明,这种饮食对S-腺苷同型半胱氨酸水解酶和腺苷激酶缺乏症患者也可能有益。补充S-腺苷蛋氨酸对蛋氨酸腺苷转移酶I/III缺乏症患者可能有用。本文给出的建议基于一般原则,在实践中应根据患者年龄、疾病严重程度、临床和实验室检查结果进行个体化调整。