Department of Chemistry, Temple University , 250B Beury Hall, 1901 North 13th Street, Philadelphia, Pennsylvania 19122, United States.
J Am Chem Soc. 2016 Nov 16;138(45):14880-14889. doi: 10.1021/jacs.6b06449. Epub 2016 Nov 8.
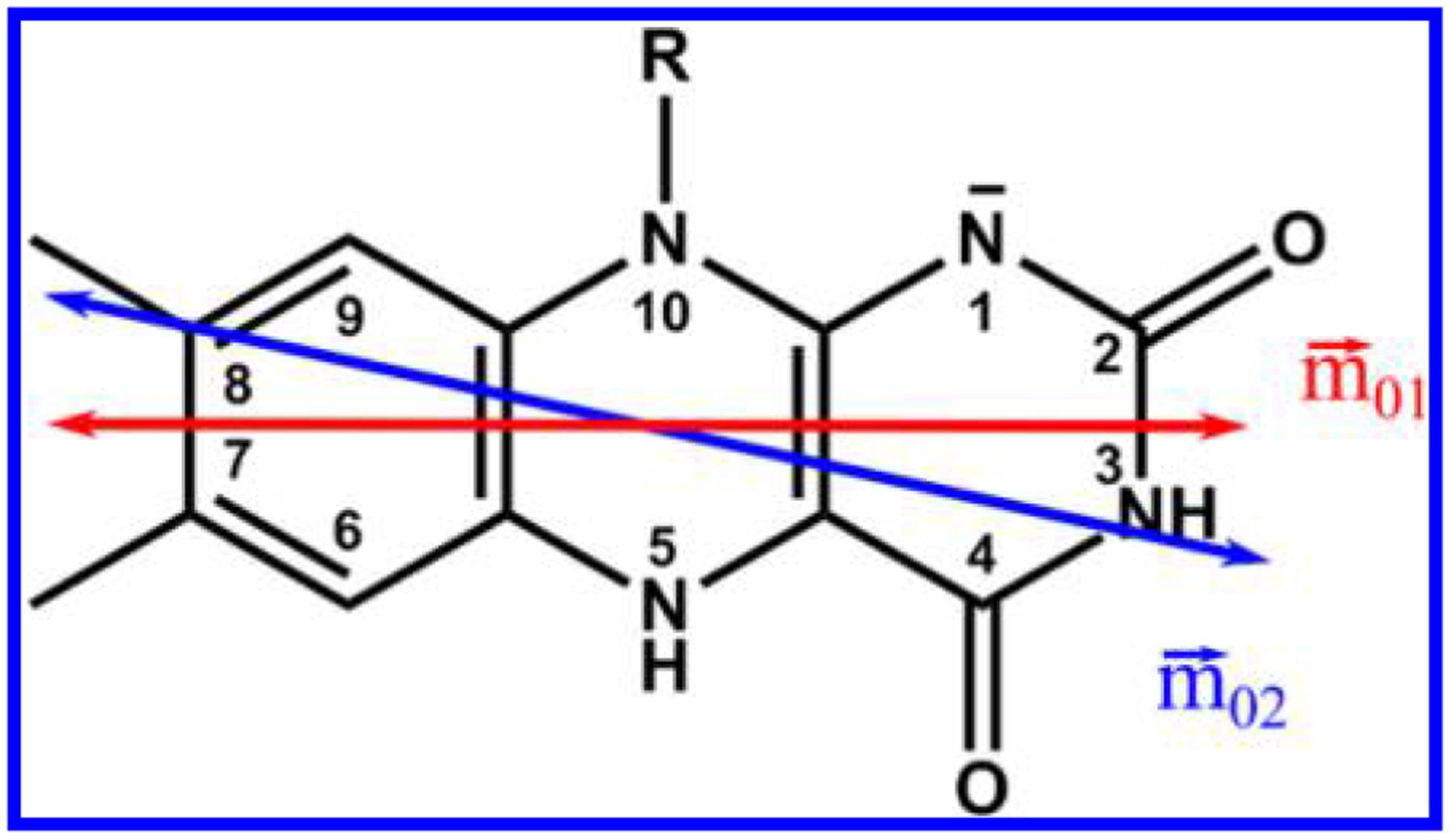
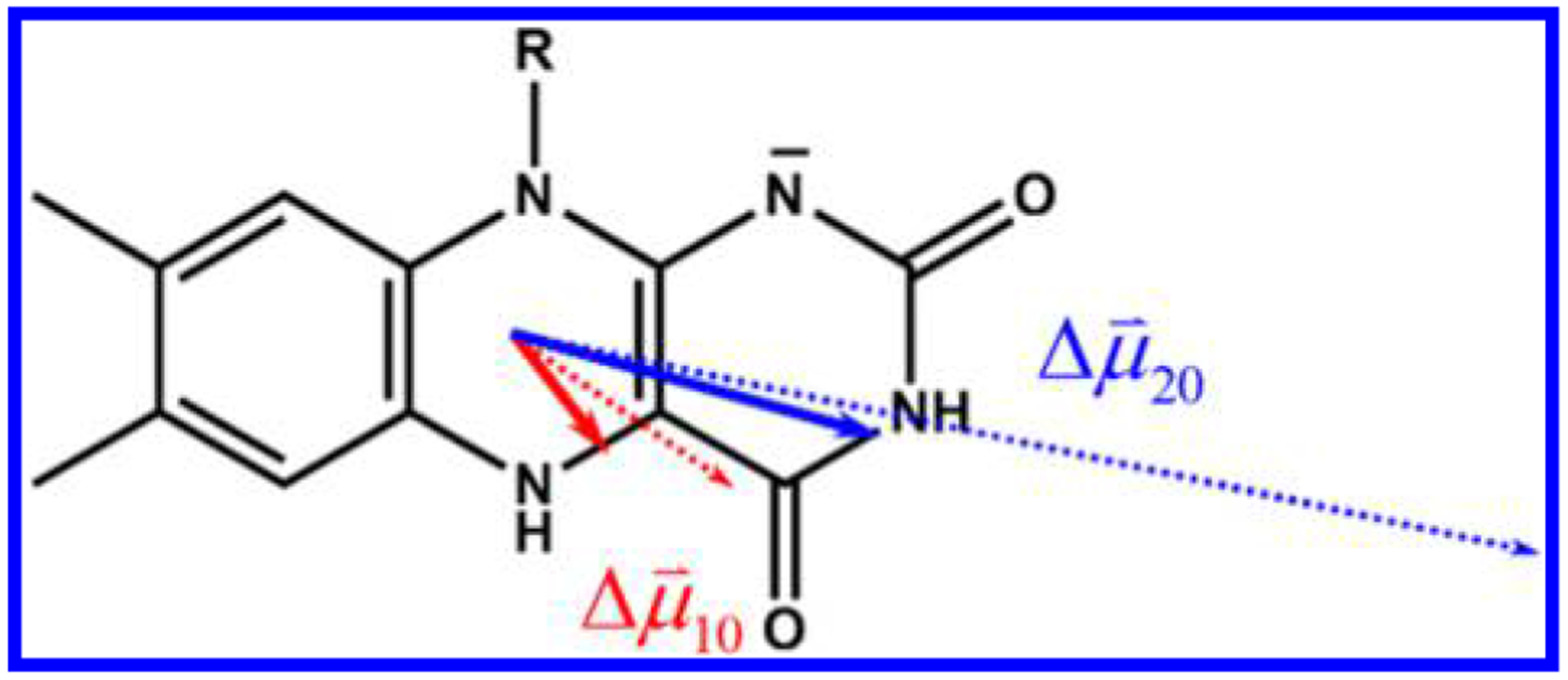
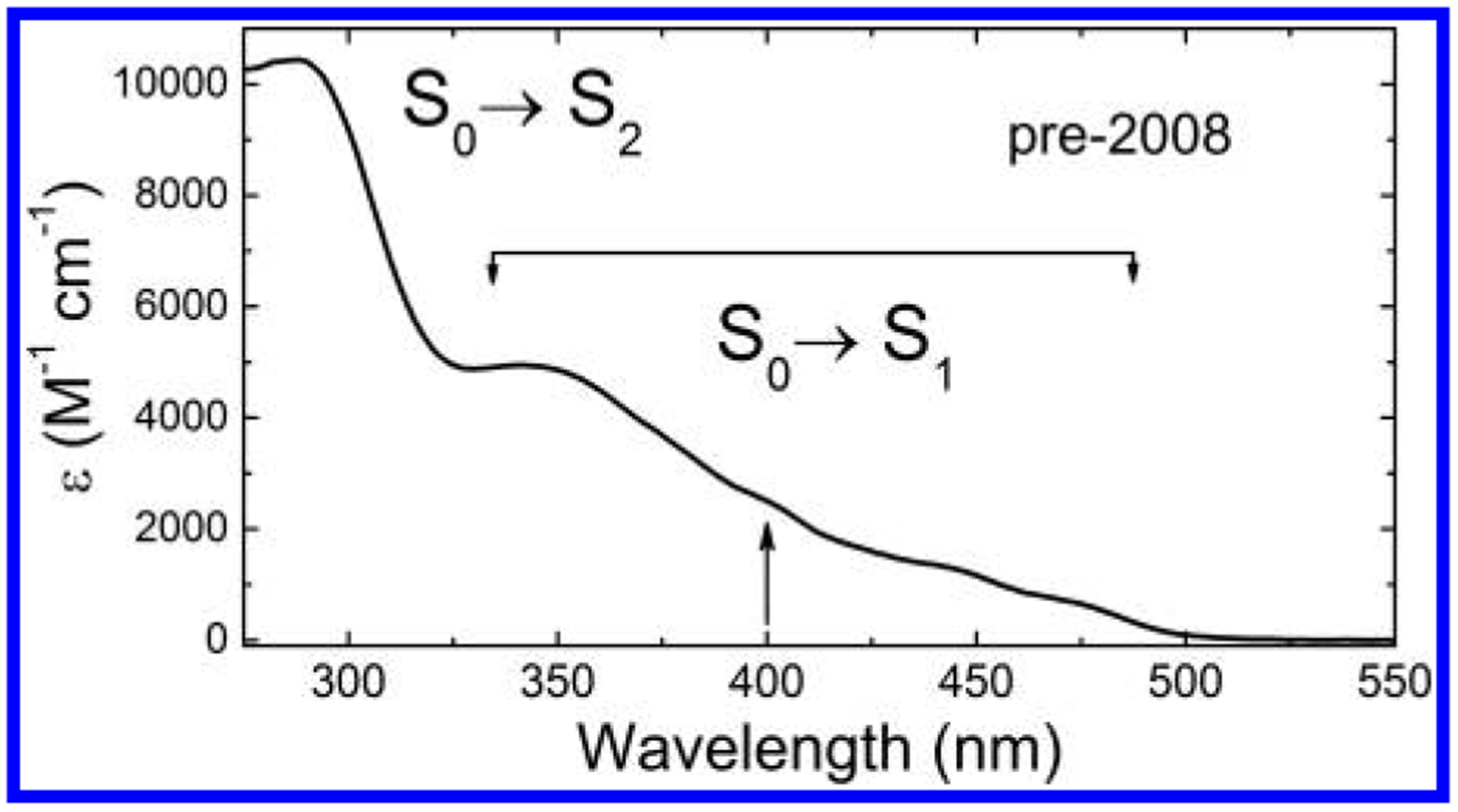
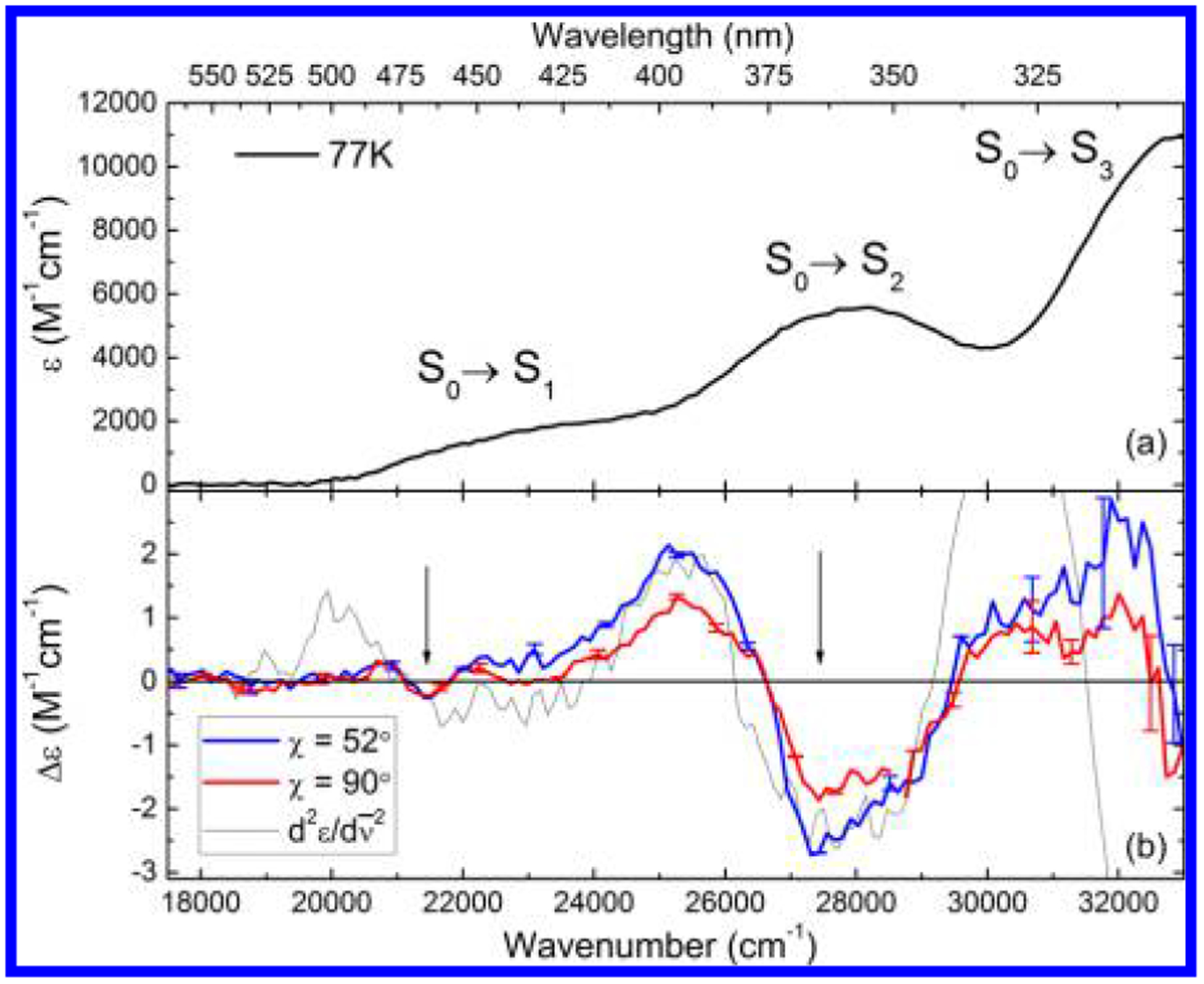
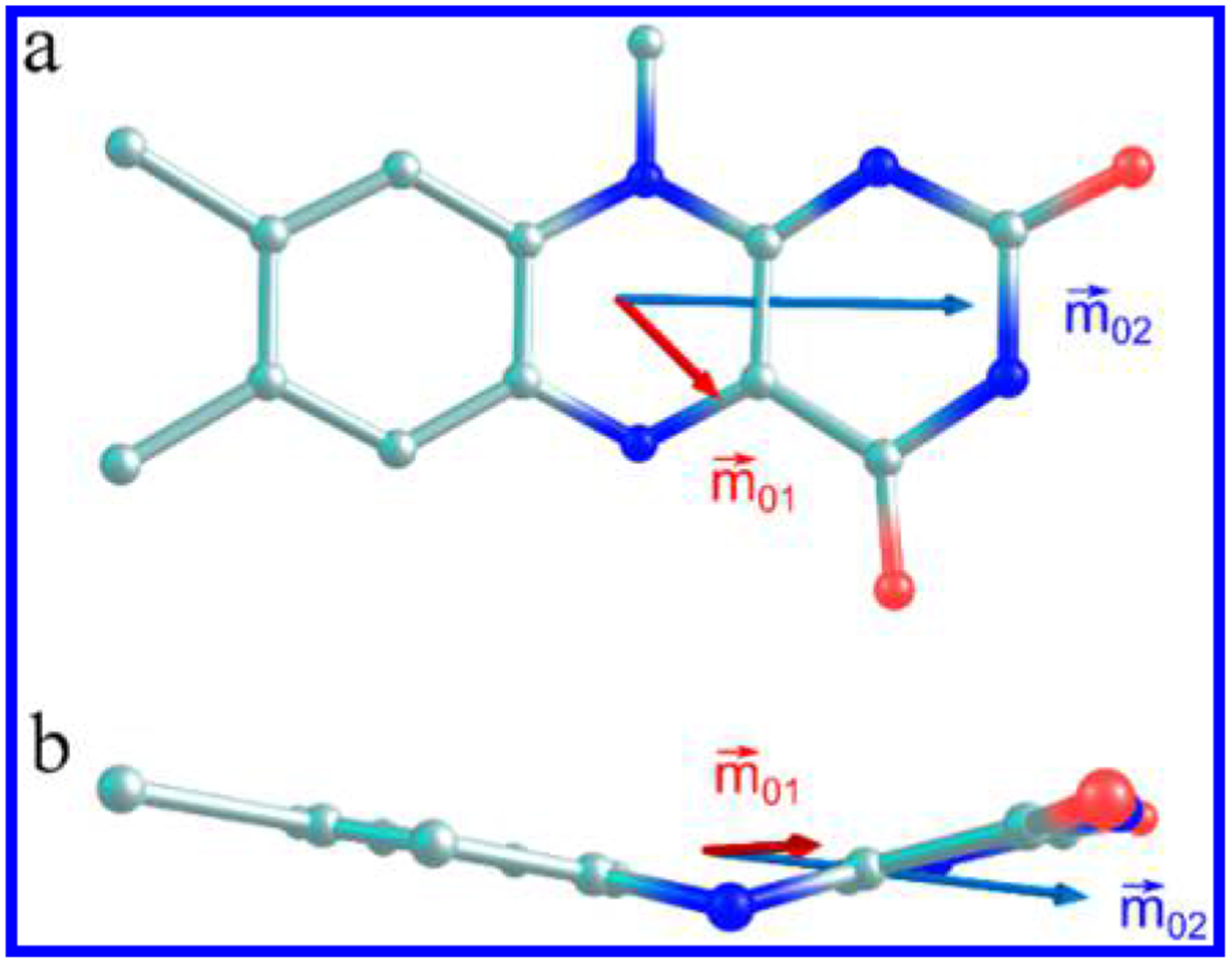
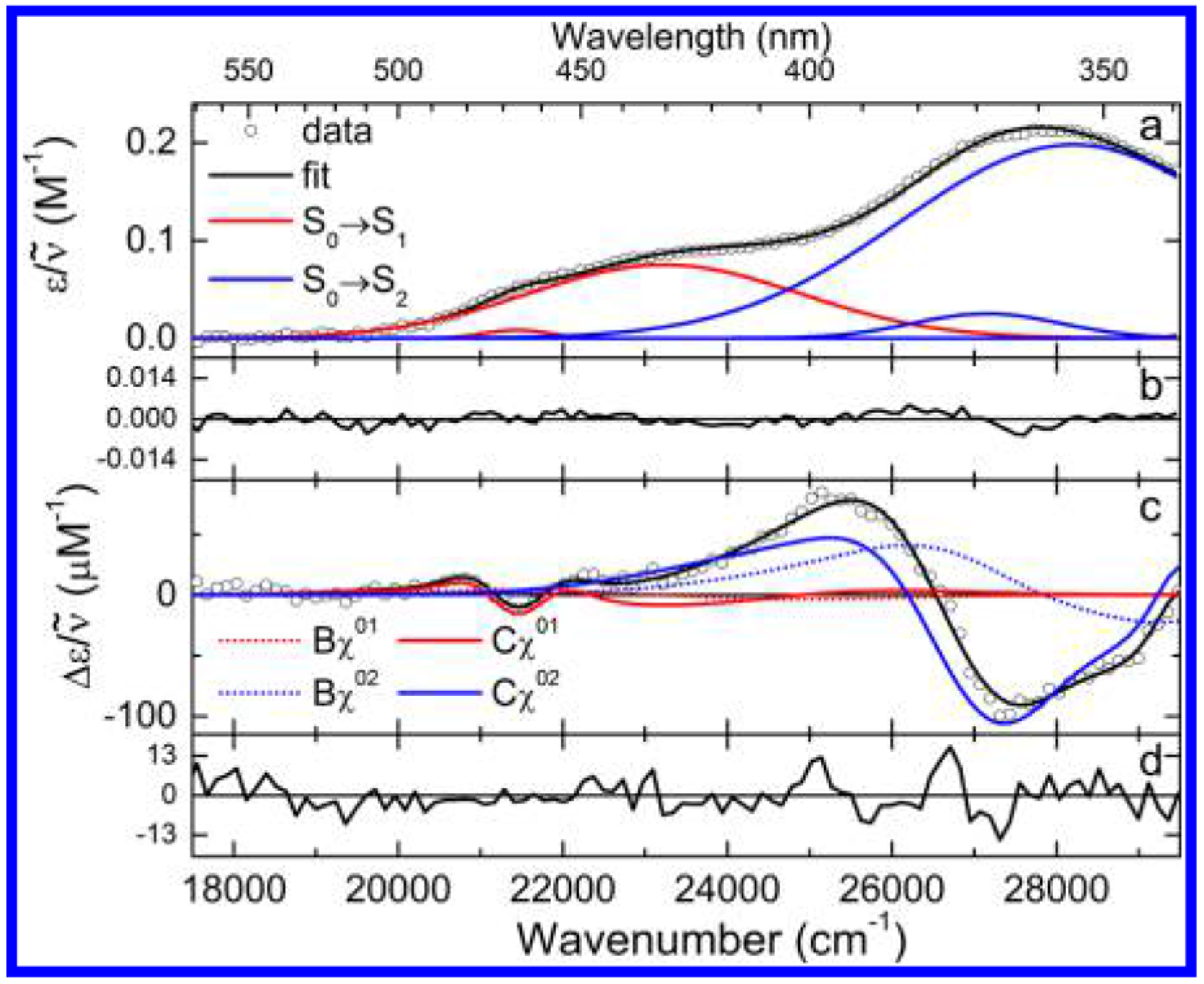
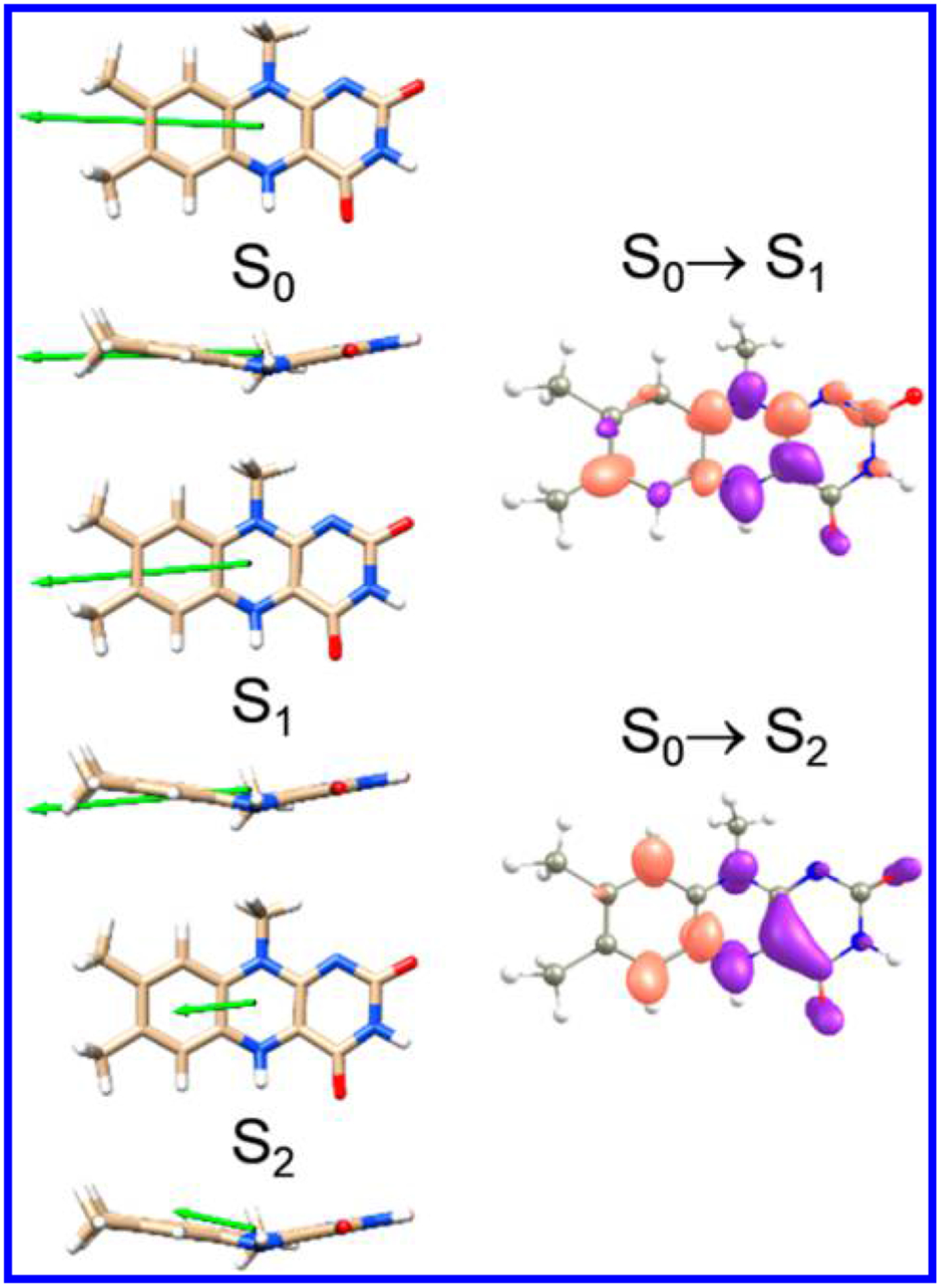
Chromophoric biomolecules are exploited as reporters of a diverse set of phenomena, acting as internal distance monitors, environment and redox sensors, and endogenous imaging probes. The extent to which they can be exploited is dependent on an accurate knowledge of their fundamental electronic properties. Arguably of greatest importance is a precise knowledge of the direction(s) of the absorption transition dipole moment(s) (TDMs) in the molecular frame of reference. Such is the case for flavins, fluorescent redox cofactors utilized for ground- and excited-state redox and photochemical processes. The directions of the TDMs in oxidized and semiquinone flavins were characterized decades ago, and the details of charge redistribution in these forms have also been studied by Stark spectroscopy. The electronic structure of the fully reduced hydroquinone anionic state, FlH, however, has been the subject of unfounded assumptions and estimates about the number and direction of TDMs in FlH, as well the electronic structure changes that occur upon light absorption. Here we have used Stark spectroscopy to measure the magnitude and direction of charge redistribution in FlH upon optical excitation. These data were analyzed using TD-DFT calculations. The results show unequivocally that not one but two nearly orientation-degenerate electronic transitions are required to explain the 340-500 nm absorption spectral range, demolishing the commonly held assumption of a single transition. The difference dipole moments for these states show that electron density shifts toward the xylene ring for both transitions. These measurements force a reappraisal of previous studies that have used erroneous assumptions and unsubstantiated estimates of these quantities. The results put future optical studies of reduced flavins/flavoproteins on a firm photophysical footing.
生色生物分子被用作各种现象的报告分子,充当内部距离监测器、环境和氧化还原传感器以及内源性成像探针。它们的可利用程度取决于对其基本电子性质的准确了解。可以说,最重要的是准确了解分子参考框架中吸收跃迁偶极矩(TDM)的方向。对于黄素来说就是如此,它是用于基态和激发态氧化还原和光化学过程的荧光氧化还原辅因子。氧化和半醌黄素的 TDM 方向几十年前就已经确定,这些形式中的电荷重新分布的细节也已经通过 Stark 光谱进行了研究。然而,完全还原的氢醌阴离子态 FlH 的电子结构一直是关于 FlH 中 TDM 的数量和方向以及光吸收时发生的电子结构变化的毫无根据的假设和估计的主题。在这里,我们使用 Stark 光谱测量了 FlH 在光激发下电荷重新分布的大小和方向。这些数据使用 TD-DFT 计算进行了分析。结果明确表明,需要两个几乎取向简并的电子跃迁才能解释 340-500nm 的吸收光谱范围,从而推翻了单一跃迁的常见假设。这些状态的差分偶极矩表明,两个跃迁的电子密度都向二甲苯环转移。这些测量迫使人们重新评估以前使用错误假设和未经证实的这些数量的估计值的研究。结果为还原黄素/黄素蛋白的未来光学研究奠定了坚实的光物理基础。