Key Laboratory for Neuro-Information of Ministry of Education, School of Life Science and Technology, Center for Informational Biology, University of Electronic Science and Technology of China, Chengdu 610054, China.
Department of Pathophysiology, Southwest Medical University, Luzhou 646000, China.
Sci Rep. 2016 Oct 4;6:34817. doi: 10.1038/srep34817.
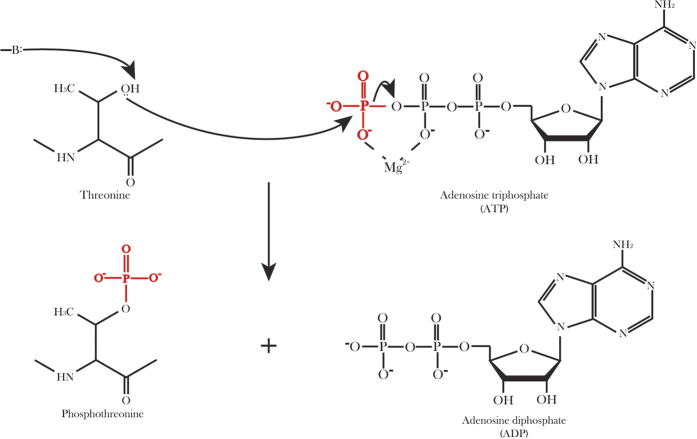
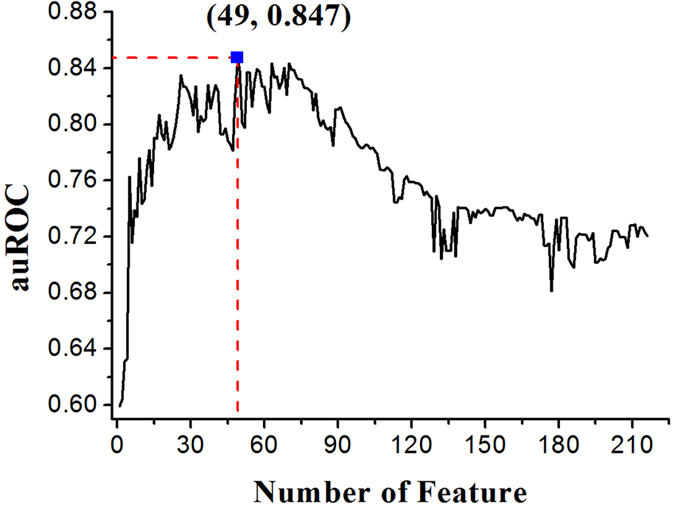
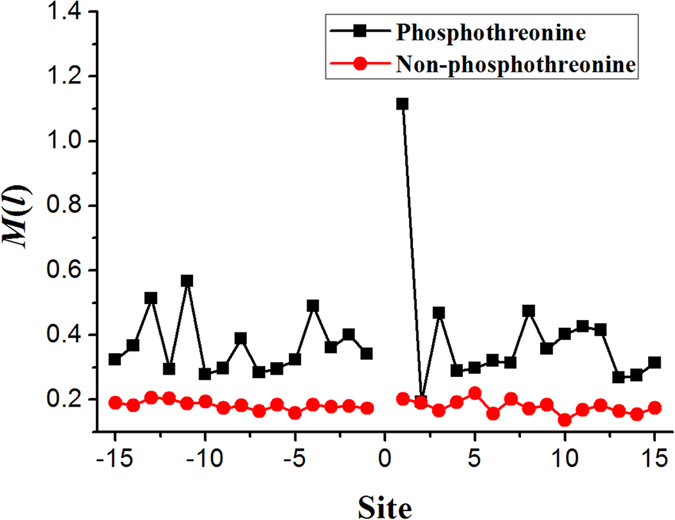
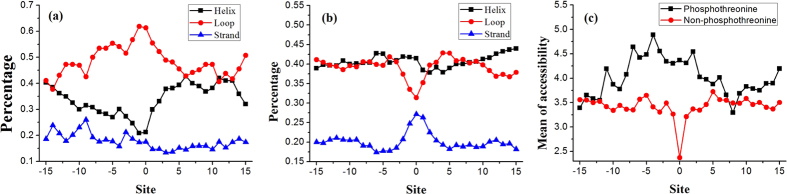
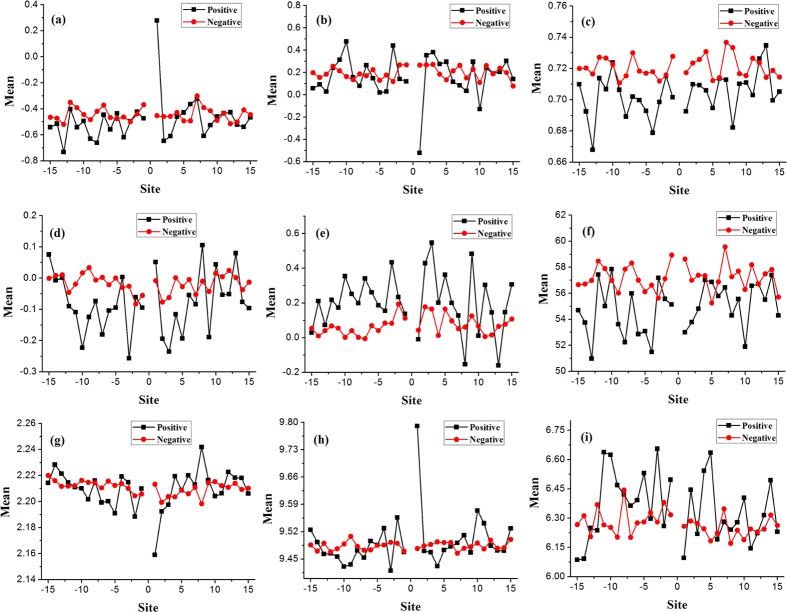
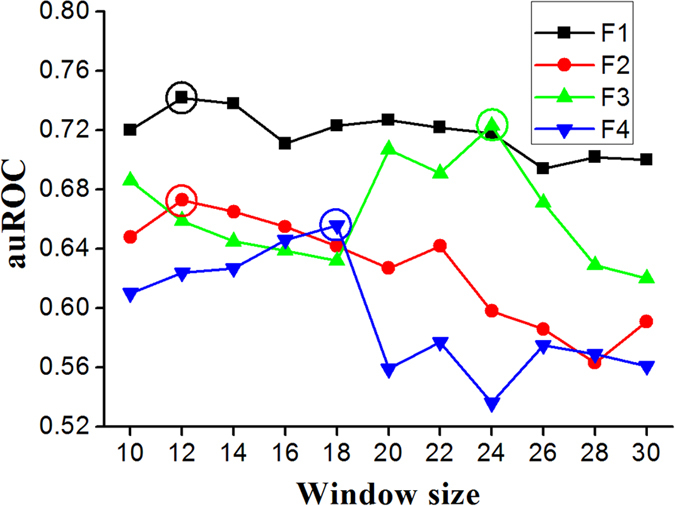
Phosphorylation is one of the most important protein post-translation modifications. With the rapid development of high-throughput mass spectrometry, phosphorylation site data is rapidly accumulating, which provides us an opportunity to systematically investigate and predict phosphorylation in proteins. The phosphorylation of threonine is the addition of a phosphoryl group to its polar side chains group. In this work, we statistically analyzed the distribution of the different properties including position conservation, secondary structure, accessibility and some other physicochemical properties of the residues surrounding the phosphothreonine site and non-phosphothreonine site. We found that the distributions of those features are non-symmetrical. Based on the distribution of properties, we developed a new model by using optimal window size strategy and feature selection technique. The cross-validated results show that the area under receiver operating characteristic curve reaches to 0.847, suggesting that our model may play a complementary role to other existing methods for predicting phosphothreonine site in proteins.
磷酸化是最重要的蛋白质翻译后修饰之一。随着高通量质谱技术的快速发展,磷酸化位点数据正在迅速积累,这为我们系统地研究和预测蛋白质中的磷酸化提供了机会。苏氨酸的磷酸化是在其极性侧链基团上添加磷酸基团。在这项工作中,我们统计分析了围绕磷酸苏氨酸位点和非磷酸苏氨酸位点的残基的不同性质(包括位置保守性、二级结构、可及性和一些其他物理化学性质)的分布。我们发现这些特征的分布是不对称的。基于性质的分布,我们使用最优窗口大小策略和特征选择技术开发了一种新模型。交叉验证结果表明,接收者操作特征曲线下的面积达到 0.847,这表明我们的模型可能对其他现有方法在蛋白质中预测磷酸苏氨酸位点起到补充作用。