Casali Nicola, Broda Agnieszka, Harris Simon R, Parkhill Julian, Brown Timothy, Drobniewski Francis
Department of Infectious Diseases and Immunity, Imperial College London, London, United Kingdom.
Centre for Immunology and Infectious Disease, Blizard Institute, Queen Mary University of London, London, United Kingdom.
PLoS Med. 2016 Oct 4;13(10):e1002137. doi: 10.1371/journal.pmed.1002137. eCollection 2016 Oct.
A large isoniazid-resistant tuberculosis outbreak centred on London, United Kingdom, has been ongoing since 1995. The aim of this study was to investigate the power and value of whole genome sequencing (WGS) to resolve the transmission network compared to current molecular strain typing approaches, including analysis of intra-host diversity within a specimen, across body sites, and over time, with identification of genetic factors underlying the epidemiological success of this cluster.
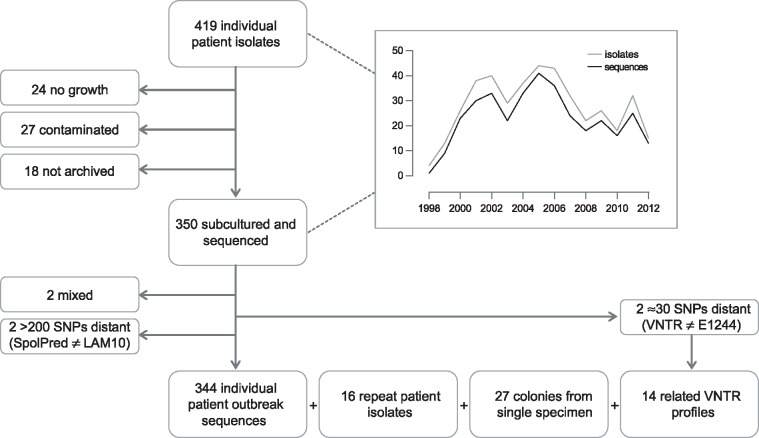
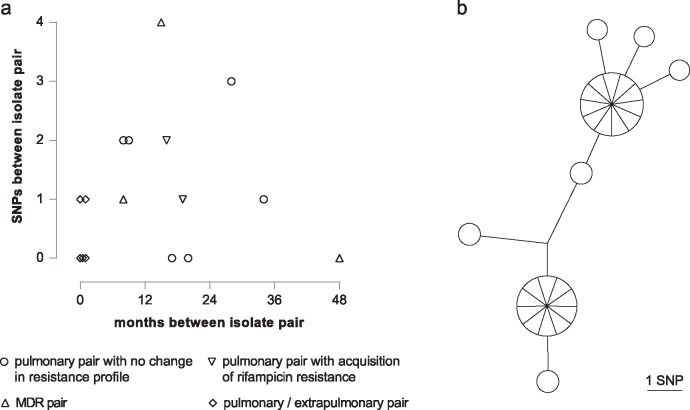
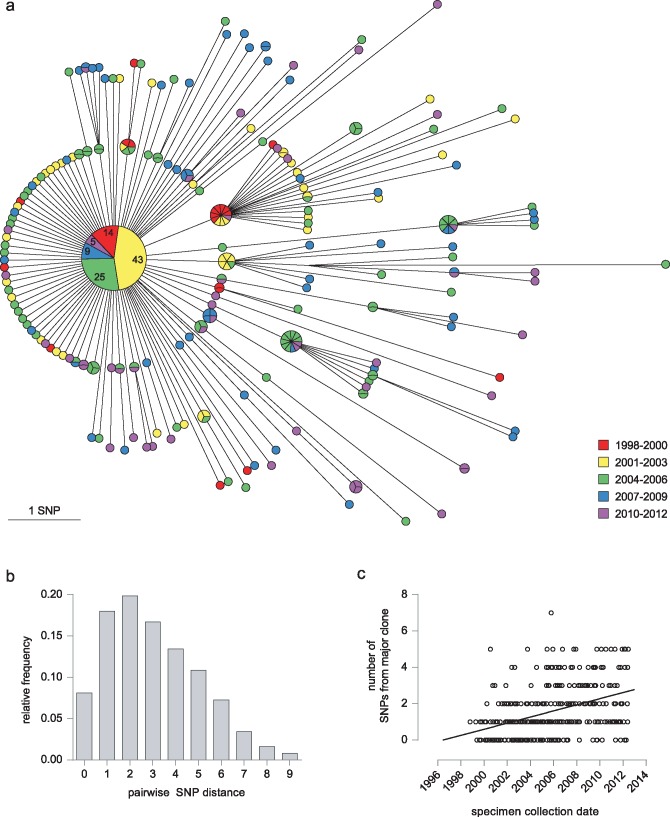
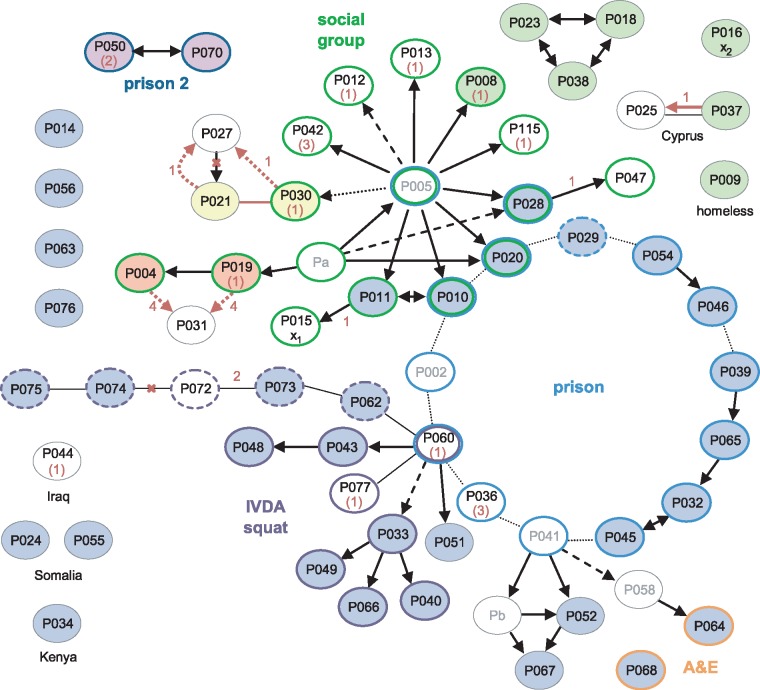
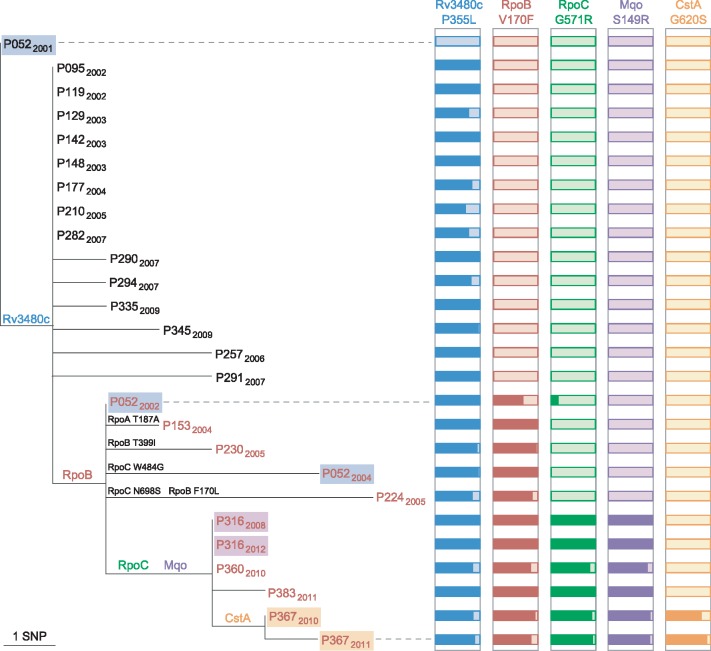
We sequenced 344 outbreak isolates from individual patients collected over 14 y (2 February 1998-22 June 2012). This demonstrated that 96 (27.9%) were indistinguishable, and only one differed from this major clone by more than five single nucleotide polymorphisms (SNPs). The maximum number of SNPs between any pair of isolates was nine SNPs, and the modal distance between isolates was two SNPs. WGS was able to reveal the direction of transmission of tuberculosis in 16 cases within the outbreak (4.7%), including within a multidrug-resistant cluster that carried a rare rpoB mutation associated with rifampicin resistance. Eleven longitudinal pairs of patient pulmonary isolates collected up to 48 mo apart differed from each other by between zero and four SNPs. Extrapulmonary dissemination resulted in acquisition of a SNP in two of five cases. WGS analysis of 27 individual colonies cultured from a single patient specimen revealed ten loci differed amongst them, with a maximum distance between any pair of six SNPs. A limitation of this study, as in previous studies, is that indels and SNPs in repetitive regions were not assessed due to the difficulty in reliably determining this variation.
Our study suggests that (1) certain paradigms need to be revised, such as the 12 SNP distance as the gold standard upper threshold to identify plausible transmissions; (2) WGS technology is helpful to rule out the possibility of direct transmission when isolates are separated by a substantial number of SNPs; (3) the concept of a transmission chain or network may not be useful in institutional or household settings; (4) the practice of isolating single colonies prior to sequencing is likely to lead to an overestimation of the number of SNPs between cases resulting from direct transmission; and (5) despite appreciable genomic diversity within a host, transmission of tuberculosis rarely results in minority variants becoming dominant. Thus, whilst WGS provided some increased resolution over variable number tandem repeat (VNTR)-based clustering, it was insufficient for inferring transmission in the majority of cases.
自1995年以来,以英国伦敦为中心爆发了大规模耐异烟肼结核病疫情。本研究的目的是调查全基因组测序(WGS)相对于当前分子菌株分型方法在解析传播网络方面的能力和价值,包括分析标本内、不同身体部位以及不同时间的宿主内多样性,同时鉴定该菌群在流行病学上成功的遗传因素。
我们对14年间(1998年2月2日至2012年6月22日)从个体患者收集的344株疫情分离株进行了测序。结果表明,96株(27.9%)无法区分,只有1株与这个主要克隆的单核苷酸多态性(SNP)差异超过5个。任意两株分离株之间的SNP最大数量为9个,分离株之间的典型距离为2个SNP。WGS能够揭示疫情中16例(4.7%)结核病的传播方向,包括在一个携带与利福平耐药相关的罕见rpoB突变的耐多药菌群中。11对纵向收集的患者肺部分离株,间隔时间长达48个月,彼此之间的SNP差异为0至4个。肺外播散导致5例中有2例获得了1个SNP。对从单个患者标本培养的27个单菌落进行WGS分析发现,它们之间有10个位点不同,任意两个之间的最大距离为6个SNP。与之前的研究一样,本研究的一个局限性是,由于难以可靠地确定重复区域的插入缺失和SNP,因此未对其进行评估。
我们的研究表明:(1)某些范式需要修订,例如将12个SNP的距离作为识别可能传播的金标准上限阈值;(2)当分离株之间存在大量SNP差异时,WGS技术有助于排除直接传播的可能性;(3)传播链或网络的概念在机构或家庭环境中可能并不适用;(4)测序前分离单菌落的做法可能会高估直接传播导致的病例之间的SNP数量;(5)尽管宿主内存在明显的基因组多样性,但结核病的传播很少导致少数变异体占主导地位。因此,虽然WGS比基于可变数目串联重复序列(VNTR)的聚类分析提供了更高的分辨率,但在大多数情况下仍不足以推断传播情况。