National Institute for Public Health and the Environment, Bilthoven, The Netherlands
National Institute for Public Health and the Environment, Bilthoven, The Netherlands.
J Clin Microbiol. 2018 Jan 24;56(2). doi: 10.1128/JCM.01100-17. Print 2018 Feb.
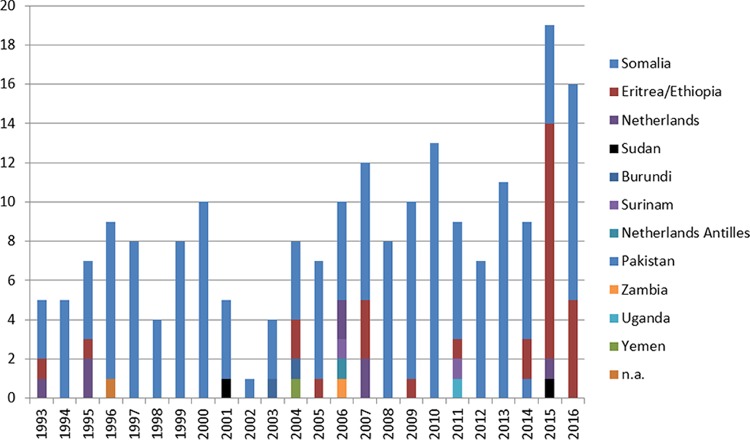
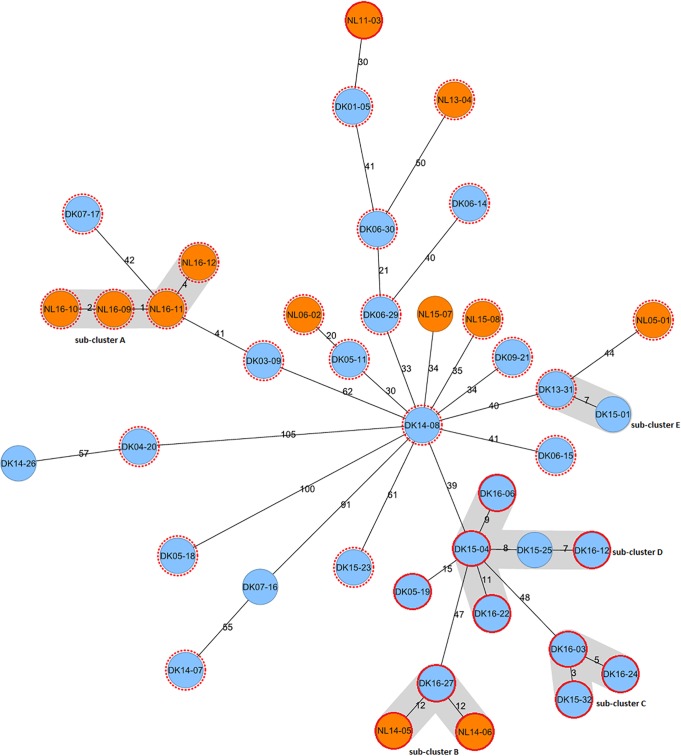
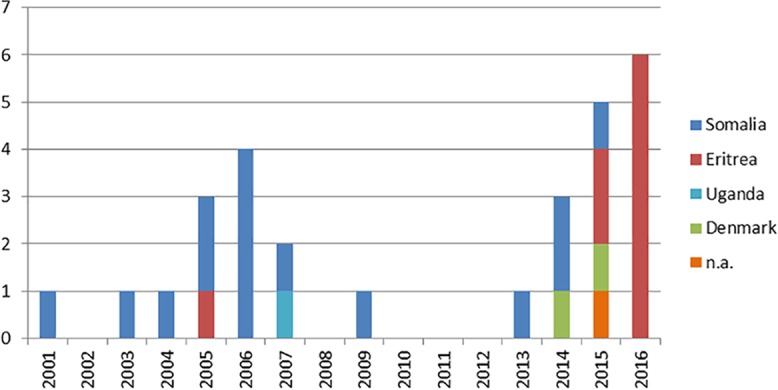
In many countries, isolates are routinely subjected to variable-number tandem-repeat (VNTR) typing to investigate transmission. Unexpectedly, cross-border clusters were identified among African refugees in the Netherlands and Denmark, although transmission in those countries was unlikely. Whole-genome sequencing (WGS) was applied to analyze transmission in depth and to assess the precision of VNTR typing. WGS was applied to 40 isolates from refugees in the Netherlands and Denmark (most of whom were from the Horn of Africa) that shared the exact same VNTR profile. Cluster investigations were undertaken to identify in-country epidemiological links. Combining WGS results for the isolates (all members of the central Asian strain [CAS]/Delhi genotype), from both European countries, an average genetic distance of 80 single-nucleotide polymorphisms (SNPs) (maximum, 153 SNPs) was observed. The few pairs of isolates with confirmed epidemiological links, except for one pair, had a maximum distance of 12 SNPs. WGS divided this refugee cluster into several subclusters of patients from the same country of origin. Although the cases, mainly originating from African countries, shared the exact same VNTR profile, most were clearly distinguished by WGS. The average genetic distance in this specific VNTR cluster was 2 times greater than that in other VNTR clusters. Thus, identical VNTR profiles did not represent recent direct transmission for this group of patients. It appears that either these strains from Africa are extremely conserved genetically or there is ongoing transmission of this genotype among refugees on their long migration routes from Africa to Europe.
在许多国家,常对 分离株进行可变数目串联重复(VNTR)分型,以调查 的传播情况。令人意外的是,在荷兰和丹麦的非洲难民中发现了跨境集群,尽管这些国家的传播不太可能。全基因组测序(WGS)被应用于深入分析传播情况,并评估 VNTR 分型的准确性。对来自荷兰和丹麦难民(其中大多数来自非洲之角)的 40 株 分离株(它们具有完全相同的 VNTR 图谱)进行了 WGS 分析。开展了集群调查,以确定国内的流行病学联系。对来自这两个欧洲国家的 分离株(均属于中亚株/德里基因型)的 WGS 结果进行组合,观察到平均 80 个单核苷酸多态性(SNP)的遗传距离(最大为 153 SNP)。除了一对之外,除了一对之外,证实具有流行病学联系的几对分离株之间的最大距离为 12 SNP。WGS 将这一难民集群分为来自同一原籍国的多个亚群。尽管这些主要来自非洲国家的 病例具有完全相同的 VNTR 图谱,但大多数病例在 WGS 下都能明显区分。在这个特定的 VNTR 集群中,平均遗传距离是其他 VNTR 集群的 2 倍。因此,对于这群患者来说,相同的 VNTR 图谱并不代表最近的直接传播。似乎这些来自非洲的菌株在遗传上非常保守,或者在非洲到欧洲的漫长迁徙过程中,这种基因型在难民中持续传播。