Yoon Young Me, Storm Kelsie J, Kamimae-Lanning Ashley N, Goloviznina Natalya A, Kurre Peter
Department of Pediatrics, Oregon Health & Science University, Portland, OR 97239, USA; Papé Family Pediatric Research Institute, Oregon Health & Science University, Portland, OR 97239, USA; Pediatric Cancer Biology Program, Oregon Health & Science University, Portland, OR 97239, USA.
Department of Pediatrics, Oregon Health & Science University, Portland, OR 97239, USA; Papé Family Pediatric Research Institute, Oregon Health & Science University, Portland, OR 97239, USA; Pediatric Cancer Biology Program, Oregon Health & Science University, Portland, OR 97239, USA; OHSU Knight Cancer Institute, Oregon Health & Science University, 3181 Southwest Sam Jackson Park Road, Portland, OR 97239, USA.
Stem Cell Reports. 2016 Nov 8;7(5):840-853. doi: 10.1016/j.stemcr.2016.09.005. Epub 2016 Oct 6.
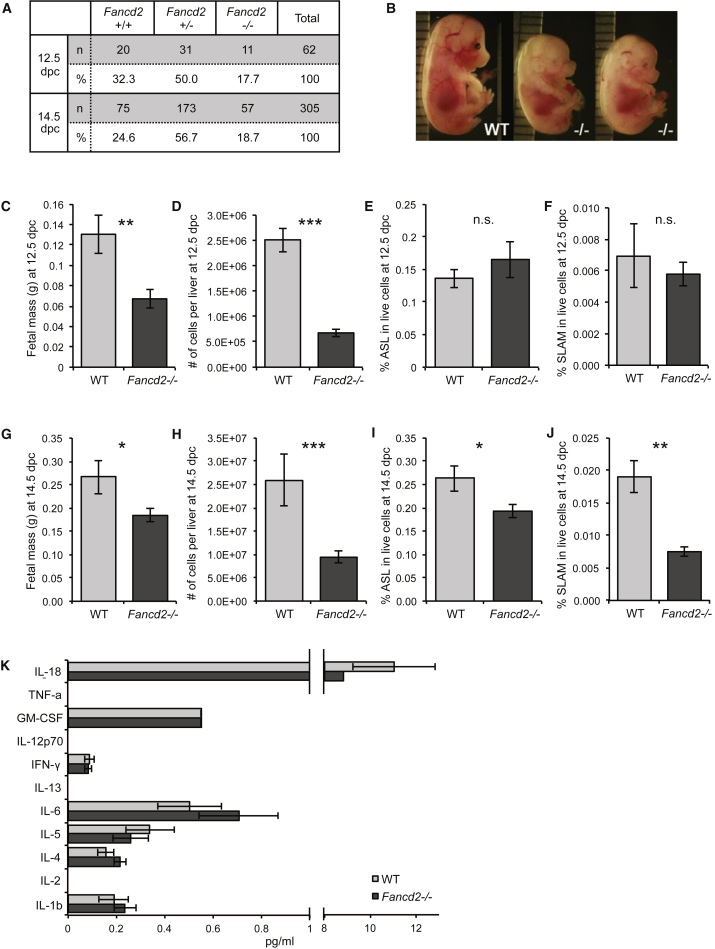
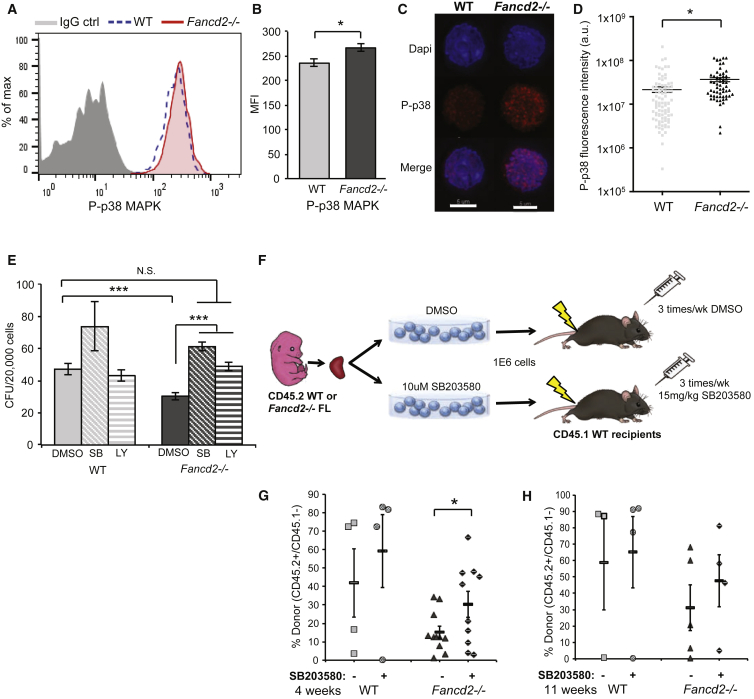
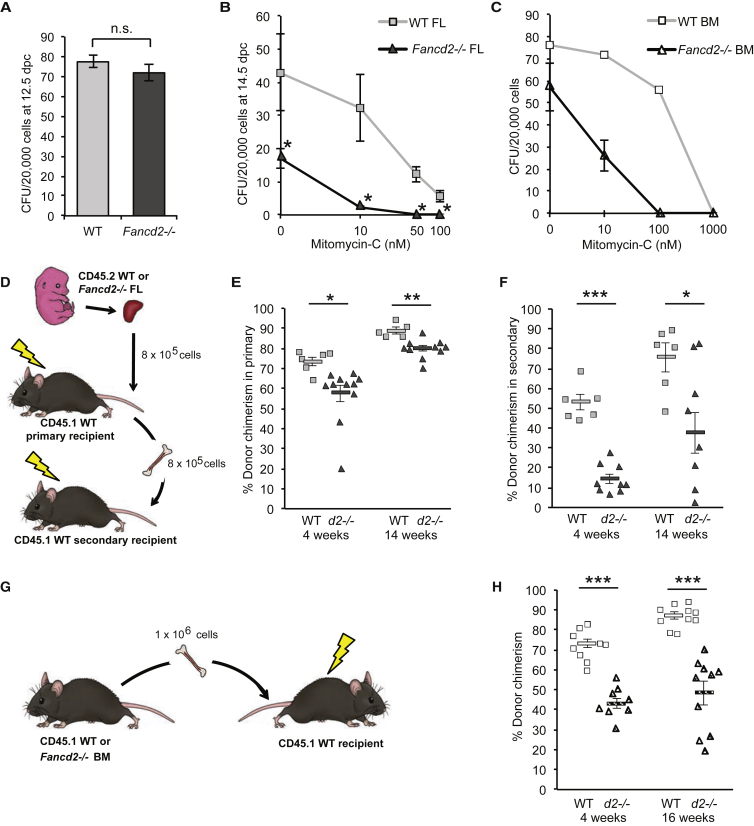
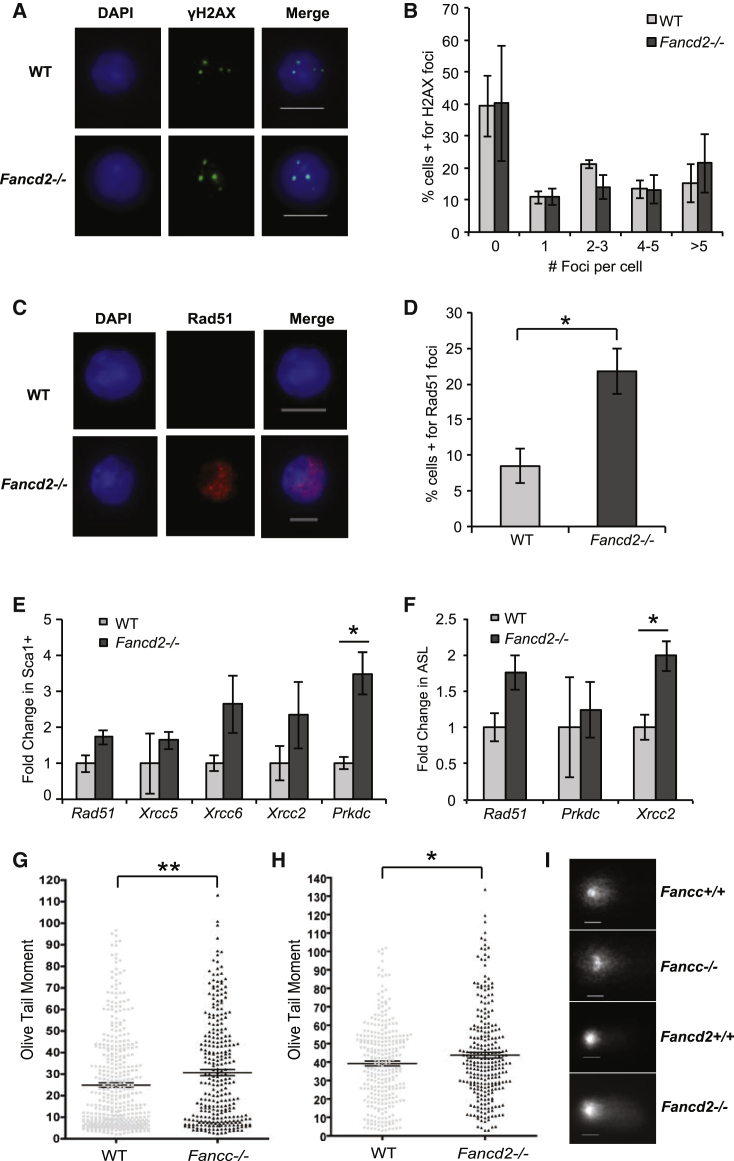
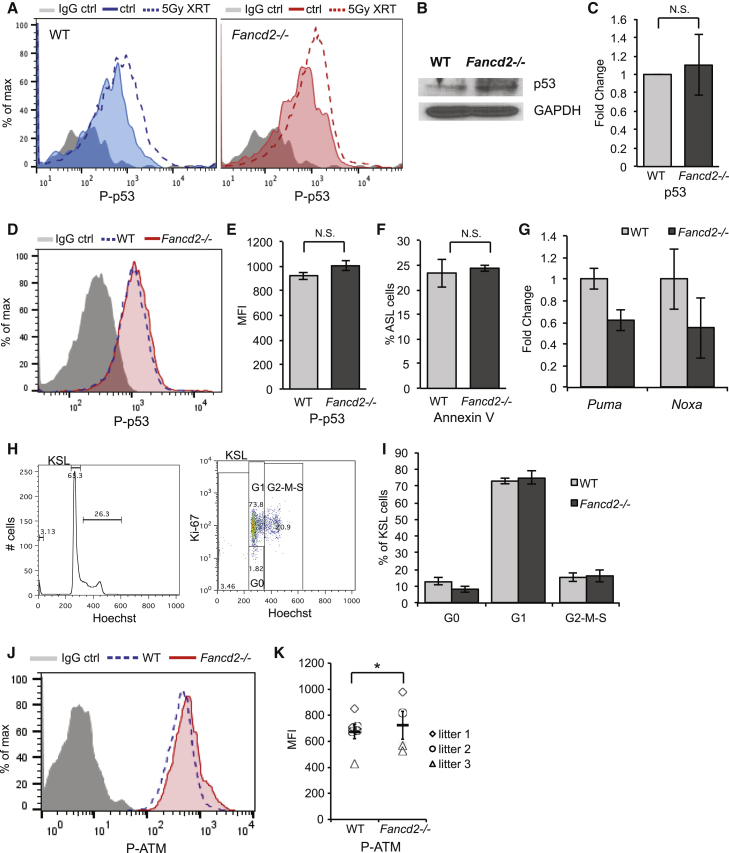
Our mechanistic understanding of Fanconi anemia (FA) pathway function in hematopoietic stem and progenitor cells (HSPCs) owes much to their role in experimentally induced DNA crosslink lesion repair. In bone marrow HSPCs, unresolved stress confers p53-dependent apoptosis and progressive cell attrition. The role of FA proteins during hematopoietic development, in the face of physiological replicative demand, remains elusive. Here, we reveal a fetal HSPC pool in Fancd2 mice with compromised clonogenicity and repopulation. Without experimental manipulation, fetal Fancd2 HSPCs spontaneously accumulate DNA strand breaks and RAD51 foci, associated with a broad transcriptional DNA-damage response, and constitutive activation of ATM as well as p38 stress kinase. Remarkably, the unresolved stress during rapid HSPC pool expansion does not trigger p53 activation and apoptosis; rather, it constrains proliferation. Collectively our studies point to a role for the FA pathway during hematopoietic development and provide a new model for studying the physiological function of FA proteins.
我们对范可尼贫血(FA)通路在造血干细胞和祖细胞(HSPCs)中的功能机制的理解,很大程度上归功于它们在实验诱导的DNA交联损伤修复中的作用。在骨髓造血干细胞中,未解决的应激会导致p53依赖的细胞凋亡和细胞逐渐损耗。面对生理复制需求时,FA蛋白在造血发育过程中的作用仍然难以捉摸。在这里,我们在Fancd2基因敲除小鼠中发现了一个克隆形成能力和再增殖能力受损的胎儿造血干细胞库。在没有实验操作的情况下,胎儿Fancd2造血干细胞会自发积累DNA链断裂和RAD51焦点,这与广泛的转录性DNA损伤反应以及ATM和p38应激激酶的组成性激活有关。值得注意的是,在快速的造血干细胞库扩张过程中未解决的应激不会触发p53激活和细胞凋亡;相反,它会限制细胞增殖。我们的研究共同表明了FA通路在造血发育中的作用,并为研究FA蛋白的生理功能提供了一个新模型。