Institute for Protein Research Osaka University, 3-2 Yamadaoka, Suita, Osaka, 565-0871, Japan.
Technology Research Association for Next Generation Natural Products Chemistry, 2-3-26 Aomi, Koto-Ku, Tokyo, 135-0064, Japan.
J Comput Chem. 2016 Dec 5;37(31):2687-2700. doi: 10.1002/jcc.24494. Epub 2016 Oct 13.
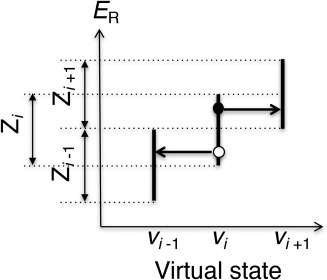
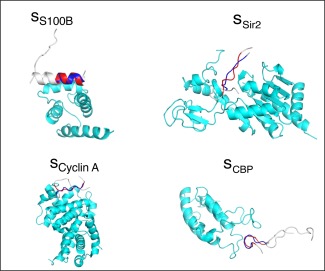
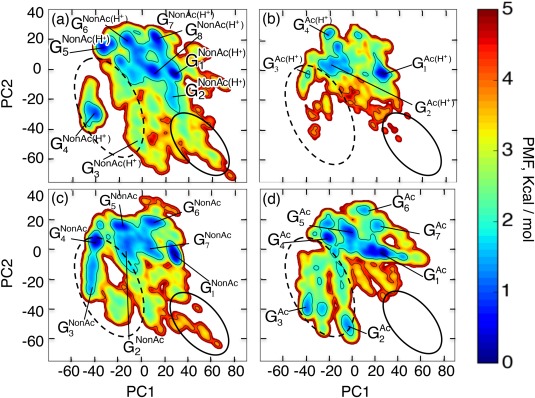
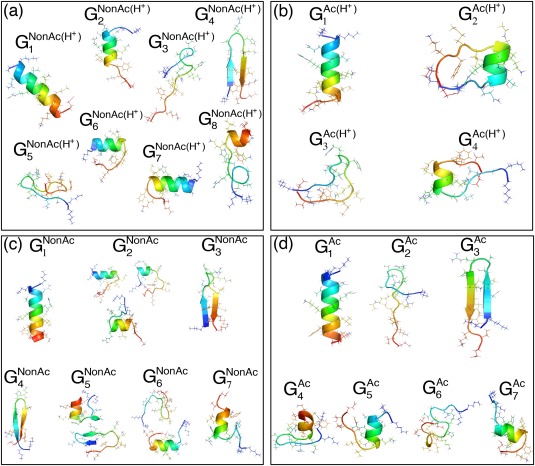
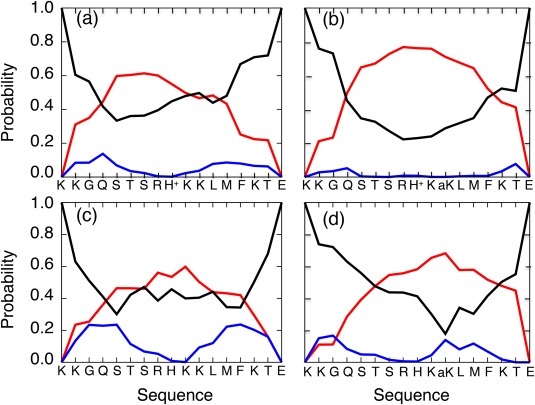
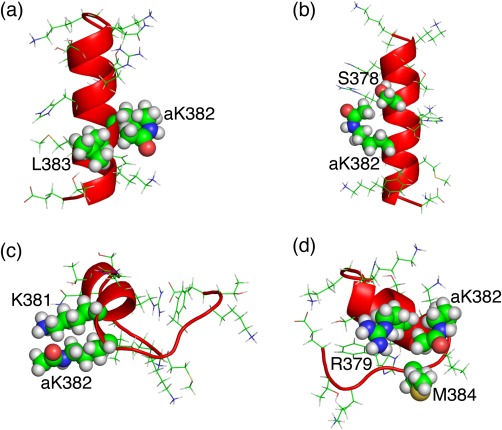
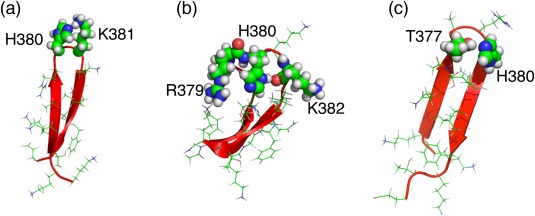
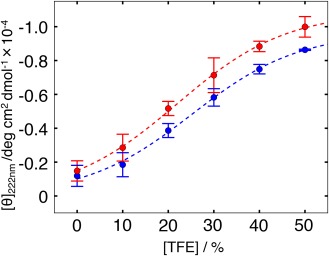
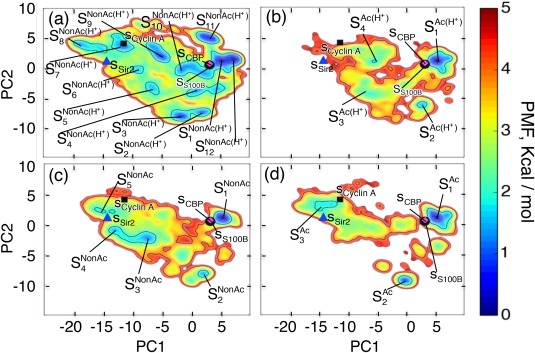
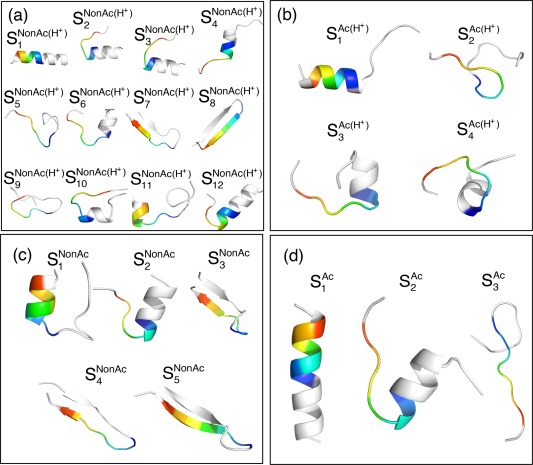
The C-terminal domain (CTD) of tumor suppressor protein p53 is an intrinsically disordered region that binds to various partner proteins, where lysine of CTD is acetylated/nonacetylated and histidine neutralized/non-neutralized. Because of the flexibility of the unbound CTD, a free-energy landscape (FEL) is a useful quantity for determining its statistical properties. We conducted enhanced conformational sampling of CTD in the unbound state via virtual system coupled multicanonical molecular dynamics, in which the lysine was acetylated or nonacetylated and histidine was charged or neutralized. The fragments were expressed by an all-atom model and were immersed in an explicit solvent. The acetylation and charge-neutralization varied FEL greatly, which might be convenient to exert a hub property. The acetylation slightly enhanced alpha-helix structures that are more compact than sheet/loop conformations. The charge-neutralization produced hairpins. Additionally, circular dichroism experiments confirmed the computational results. We propose possible binding mechanisms of CTD to partners by investigating FEL. © 2016 The Authors. Journal of Computational Chemistry Published by Wiley Periodicals, Inc.
肿瘤抑制蛋白 p53 的 C 末端结构域(CTD)是一个固有无序区域,与各种伴侣蛋白结合,其中 CTD 的赖氨酸被乙酰化/非乙酰化,组氨酸被中和/非中和。由于未结合的 CTD 的灵活性,自由能景观(FEL)是确定其统计特性的有用数量。我们通过虚拟系统耦合多正则分子动力学对未结合状态下的 CTD 进行了增强构象采样,其中赖氨酸被乙酰化或非乙酰化,组氨酸被带电荷或不带电荷。片段由全原子模型表示,并浸入显式溶剂中。乙酰化和电荷中和极大地改变了 FEL,这可能方便发挥枢纽性质。乙酰化略微增强了比片/环构象更紧凑的α-螺旋结构。电荷中和产生发夹。此外,圆二色性实验证实了计算结果。我们通过研究 FEL 提出了 CTD 与伴侣结合的可能机制。 © 2016 作者。John Wiley & Sons, Inc. 出版的《计算机化学杂志》