Cai Qian, Tammineni Prasad
J Alzheimers Dis. 2017;57(4):1087-1103. doi: 10.3233/JAD-160726.
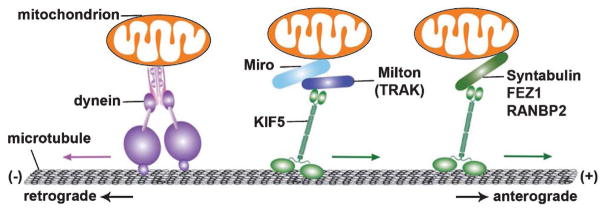
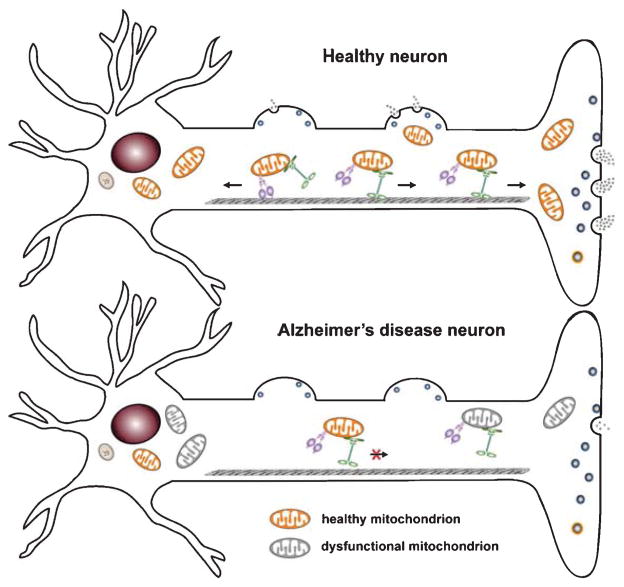
Alzheimer's disease (AD) is characterized by brain deposition of amyloid plaques and tau neurofibrillary tangles along with steady cognitive decline. Synaptic damage, an early pathological event, correlates strongly with cognitive deficits and memory loss. Mitochondria are essential organelles for synaptic function. Neurons utilize specialized mechanisms to drive mitochondrial trafficking to synapses in which mitochondria buffer Ca2+ and serve as local energy sources by supplying ATP to sustain neurotransmitter release. Mitochondrial abnormalities are one of the earliest and prominent features in AD patient brains. Amyloid-β (Aβ) and tau both trigger mitochondrial alterations. Accumulating evidence suggests that mitochondrial perturbation acts as a key factor that is involved in synaptic failure and degeneration in AD. The importance of mitochondria in supporting synaptic function has made them a promising target of new therapeutic strategies for AD. Here, we review the molecular mechanisms regulating mitochondrial function at synapses, highlight recent findings on the disturbance of mitochondrial dynamics and transport in AD, and discuss how these alterations impact synaptic vesicle release and thus contribute to synaptic pathology associated with AD.
阿尔茨海默病(AD)的特征是大脑中出现淀粉样斑块沉积和tau神经原纤维缠结,同时伴有认知功能持续下降。突触损伤是一种早期病理事件,与认知缺陷和记忆丧失密切相关。线粒体是突触功能的重要细胞器。神经元利用特殊机制驱动线粒体向突触运输,线粒体在突触中缓冲Ca2+,并通过提供ATP作为局部能量来源,以维持神经递质释放。线粒体异常是AD患者大脑中最早出现且突出的特征之一。淀粉样β蛋白(Aβ)和tau蛋白均会引发线粒体改变。越来越多的证据表明,线粒体功能紊乱是导致AD突触功能障碍和退化的关键因素。线粒体在支持突触功能方面的重要性使其成为AD新治疗策略的一个有前景的靶点。在此,我们综述调节突触线粒体功能的分子机制,强调AD中线粒体动力学和运输紊乱的最新研究发现,并讨论这些改变如何影响突触小泡释放,进而导致与AD相关的突触病理。