Sanchez-Villamil Javier, Tapia-Pastrana Gabriela, Navarro-Garcia Fernando
Department of Cell Biology, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional México City, Mexico.
Front Cell Infect Microbiol. 2016 Oct 7;6:120. doi: 10.3389/fcimb.2016.00120. eCollection 2016.
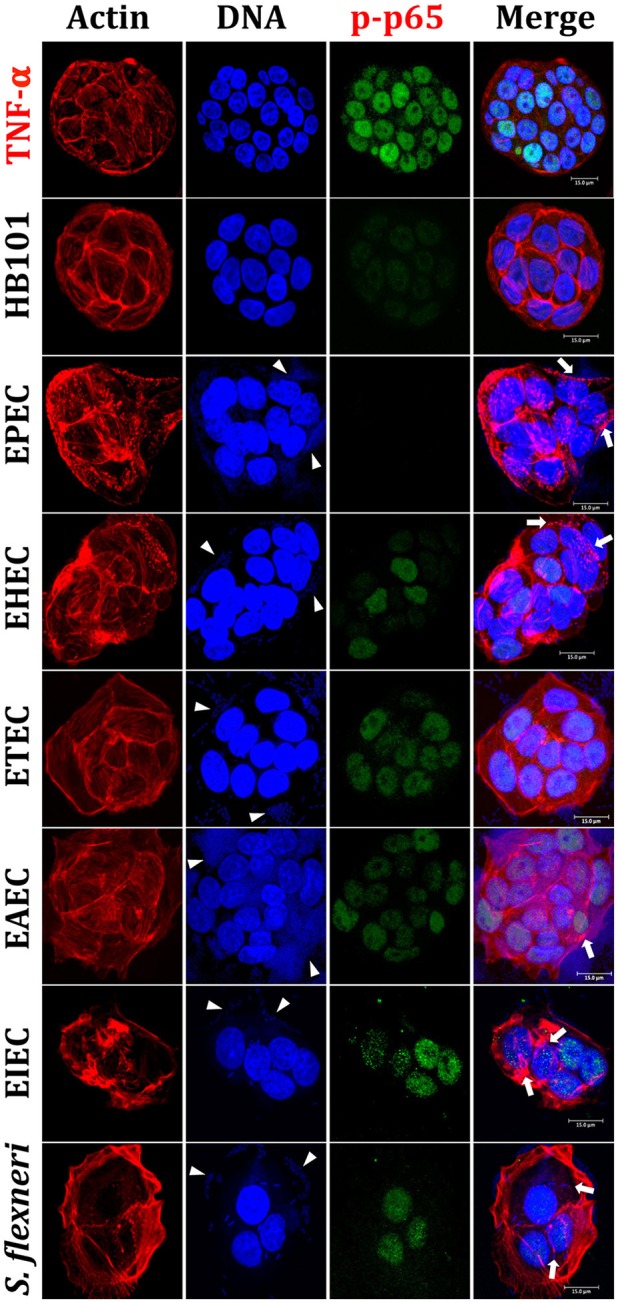
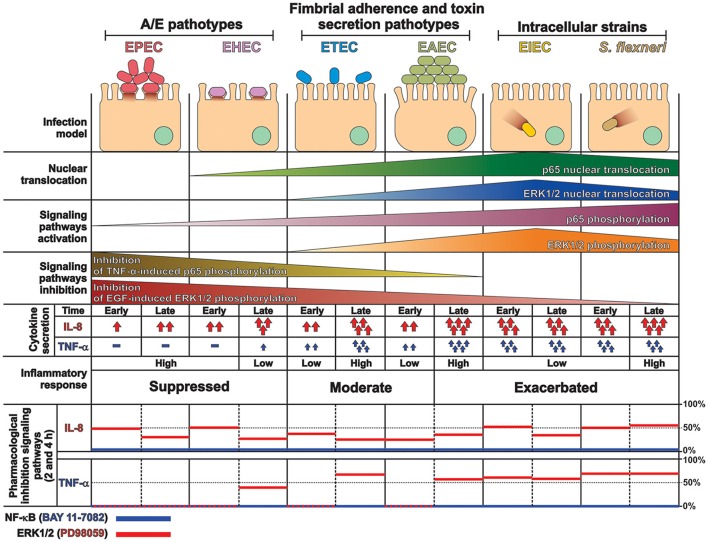
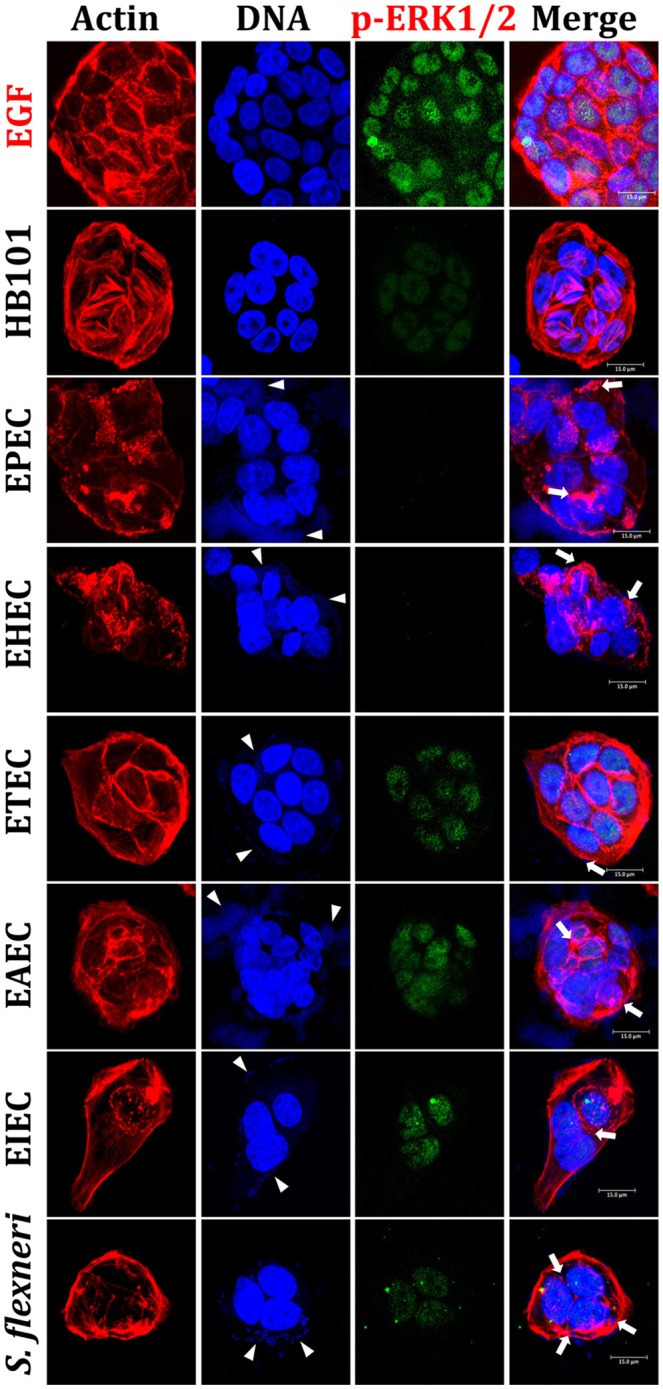
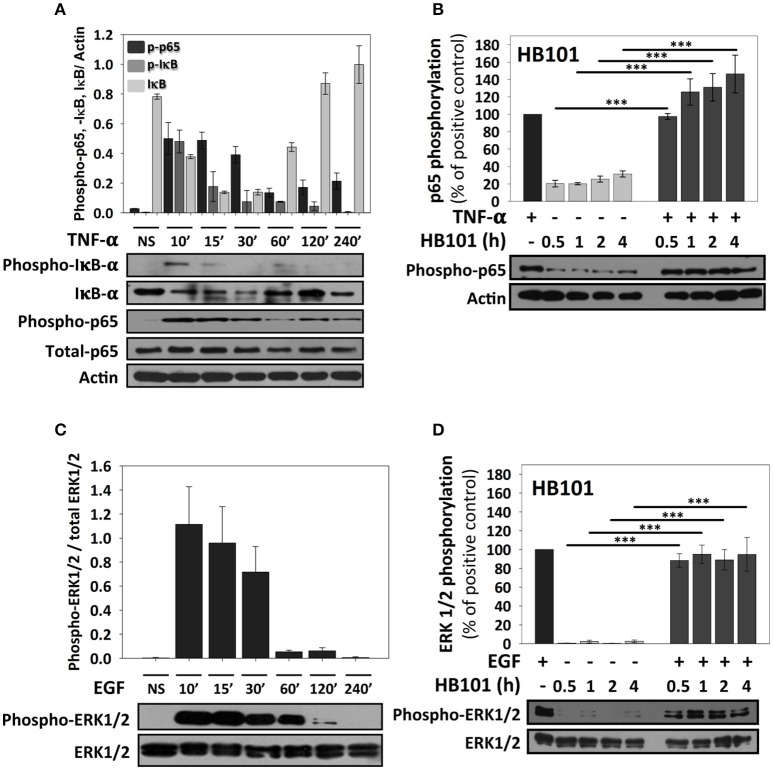
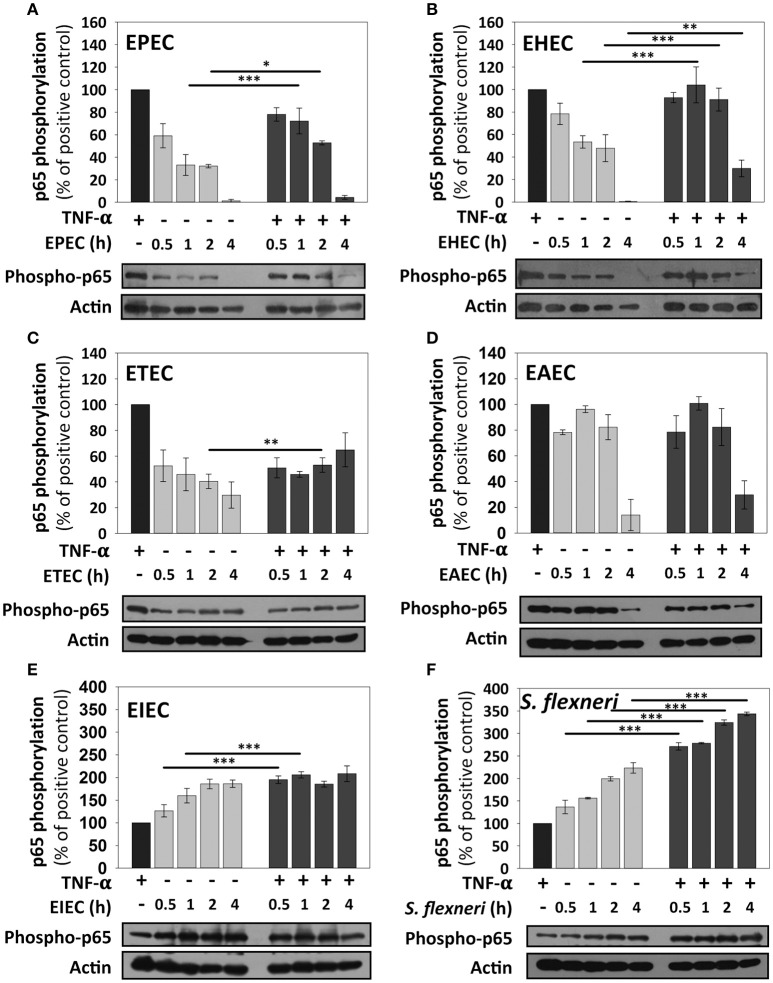
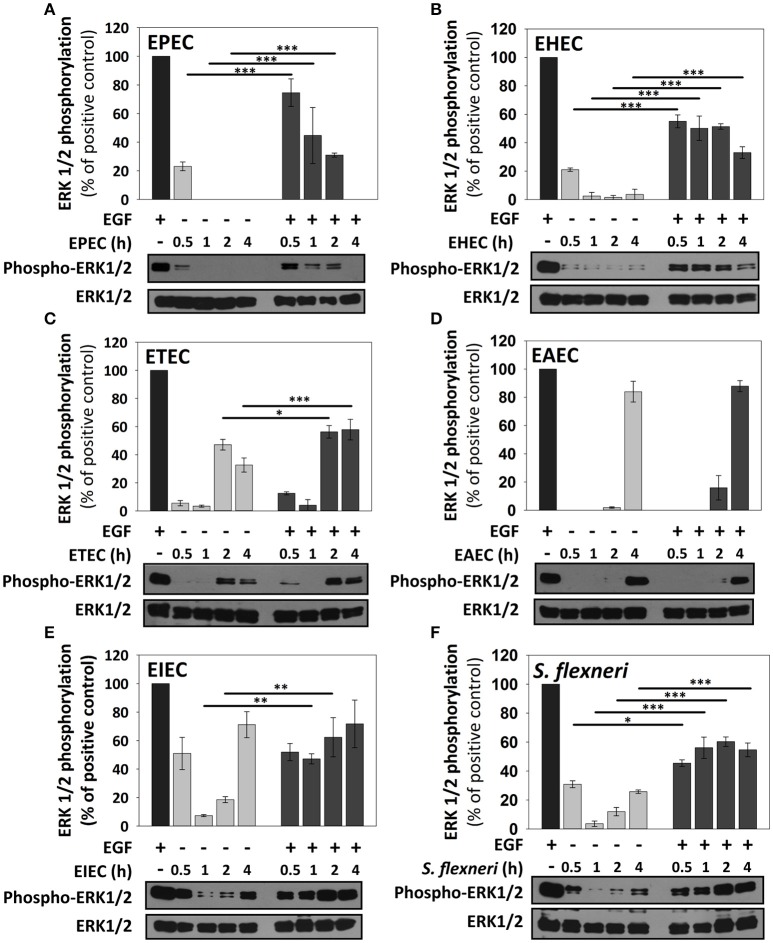
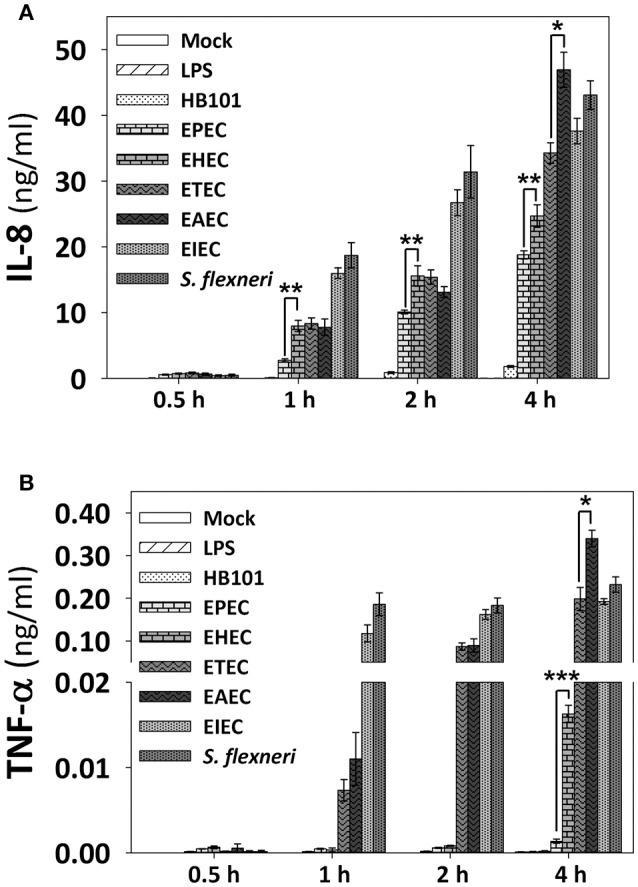
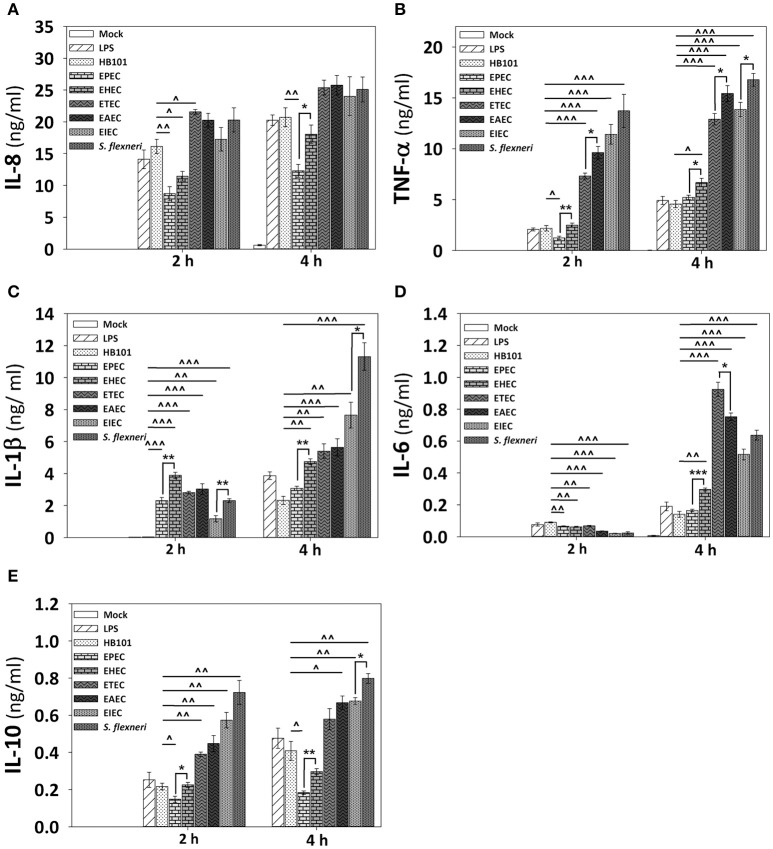
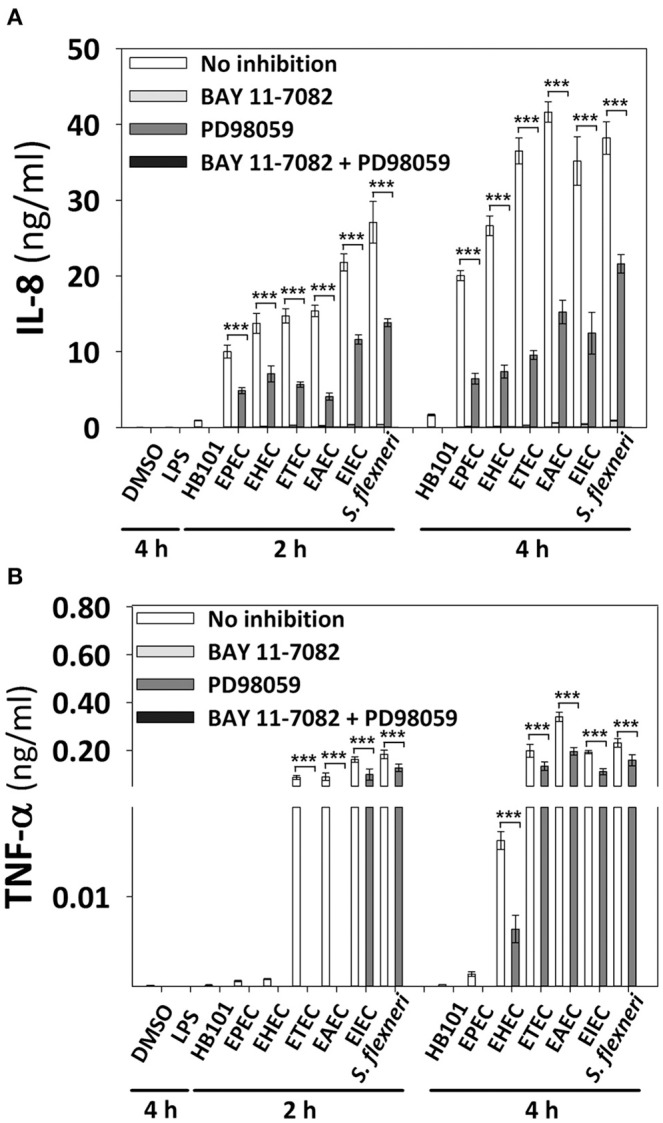
Inflammatory response is key for the host defense against diarrheagenic and contributes to the pathogenesis of the disease but there is not a comparative study among different diarrheagenic pathotypes. We analyzed the inflammatory response induced by five diarrheagenic pathotypes in a HT-29 cell infection model. The model was unified to reproduce the pathogenesis of each pathotype. To compare the inflammatory responses we evaluated: (i) nuclear NF-κB and ERK1/2 translocation by confocal microscopy; (ii) kinetics of activation by each pathway detecting p65 and ERK1/2 phosphorylation by Western blotting; (iii) pathways modulation through bacterial infections with or without co-stimulation with TNF-α or EGF; (iv) cytokine profile induced by each pathotype with and without inhibitors of each pathway. EHEC but mainly EPEC inhibited translocation and activation of p65 and ERK1/2 pathways, as well as cytokines secretion; inhibition of p65 and ERK1/2 phosphorylation prevailed in the presence of TNF-α and EGF, respectively. Intracellular strains, EIEC/, caused a strong translocation, activation, and cytokines secretion but they could not inhibit TNF-α and EGF stimulation. ETEC and mainly EAEC caused a moderate translocation, but a differential activation, and high cytokines secretion; interestingly TNF-α and EGF stimulation did no modify p65 and ERK1/2 activation. The use of inhibitors of NF-κB and/or ERK1/2 showed that NF-κB is crucial for cytokine induction by the different pathotypes; only partially depended on ERK1/2 activation. Thus, in spite of their differences, the pathotypes can also be divided in three groups according to their inflammatory response as those (i) that inject effectors to cause A/E lesion, which are able to inhibit NF-κB and ERK1/2 pathways, and cytokine secretion; (ii) with fimbrial adherence and toxin secretion with a moderate inhibition of both pathways but high cytokines secretion through autocrine cytokine regulation; and (iii) the intracellular bacteria that induce the highest pathways activation and cytokines secretion by using different activation mechanisms. This study provides a comprehensive analysis of how the different pathogenesis schemes of pathotypes manipulate inflammatory signaling pathways, which leads to a specific proinflammatory cytokine secretion in a cell model infection that reproduce the hallmarks of infection of each pathotype.
炎症反应是宿主抵御致泻性病菌的关键,且在疾病发病机制中发挥作用,但尚未有针对不同致泻性病菌型的比较研究。我们在HT - 29细胞感染模型中分析了五种致泻性病菌型诱导的炎症反应。该模型经过统一构建以再现每种病菌型的发病机制。为比较炎症反应,我们评估了:(i) 通过共聚焦显微镜观察核NF - κB和ERK1/2的易位;(ii) 通过蛋白质印迹法检测p65和ERK1/2磷酸化来分析各信号通路的激活动力学;(iii) 通过有无TNF - α或EGF共刺激的细菌感染来研究信号通路的调节;(iv) 使用各信号通路抑制剂前后各病菌型诱导的细胞因子谱。肠出血性大肠杆菌(EHEC),尤其是肠致病性大肠杆菌(EPEC),抑制p65和ERK1/2信号通路的易位与激活以及细胞因子分泌;在分别存在TNF - α和EGF时,p65和ERK1/2磷酸化的抑制作用更为明显。细胞内病菌型,如侵袭性大肠杆菌(EIEC),会引发强烈的易位、激活及细胞因子分泌,但无法抑制TNF - α和EGF刺激。产肠毒素大肠杆菌(ETEC),尤其是肠集聚性大肠杆菌(EAEC),会引发中等程度的易位,但激活情况存在差异,且细胞因子分泌量较高;有趣的是,TNF - α和EGF刺激并未改变p65和ERK1/2的激活。使用NF - κB和/或ERK1/2抑制剂表明,NF - κB对不同病菌型诱导细胞因子至关重要;仅部分依赖于ERK1/2激活。因此,尽管各病菌型存在差异,但根据其炎症反应也可分为三组:(i) 那些注入效应蛋白导致A/E损伤的病菌型,能够抑制NF - κB和ERK1/2信号通路以及细胞因子分泌;(ii) 具有菌毛黏附及毒素分泌功能,对两条信号通路均有中等程度抑制,但通过自分泌细胞因子调节实现高细胞因子分泌的病菌型;(iii) 通过不同激活机制诱导最高程度信号通路激活及细胞因子分泌的细胞内病菌型。本研究全面分析了不同病菌型的发病机制如何操控炎症信号通路,从而在重现每种病菌型感染特征的细胞模型感染中导致特定的促炎细胞因子分泌。