Leone Vanessa, Faraldo-Gómez José D
Theoretical Molecular Biophysics Section, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD 20892
J Gen Physiol. 2016 Dec;148(6):441-457. doi: 10.1085/jgp.201611679. Epub 2016 Nov 7.
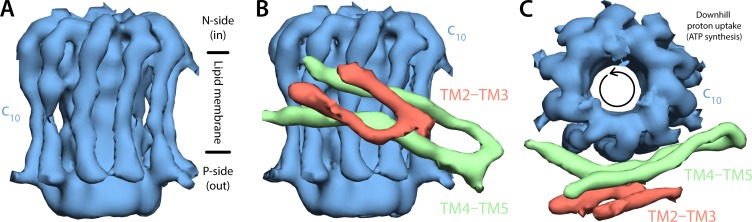
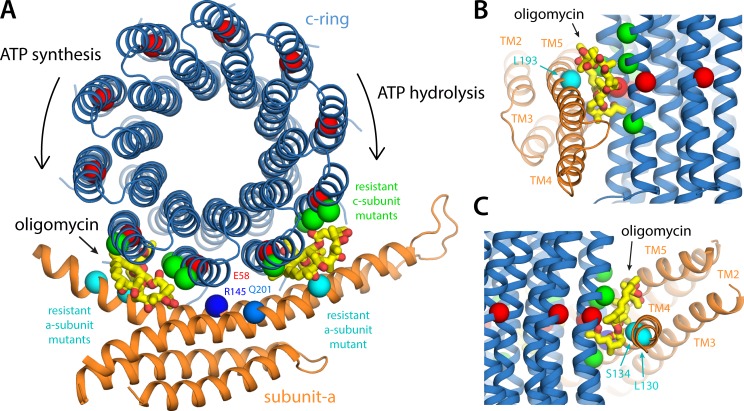
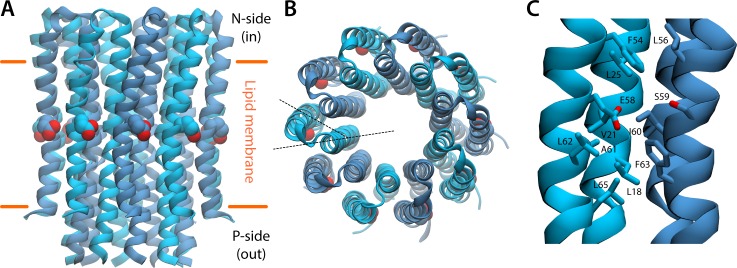
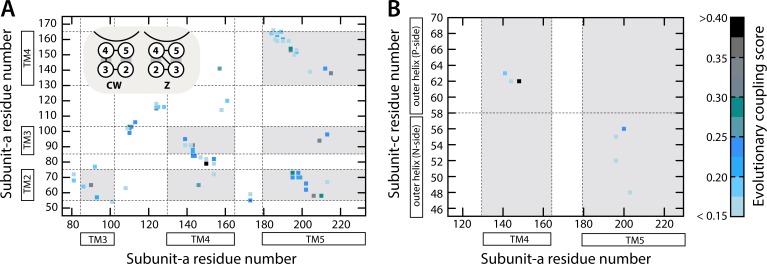
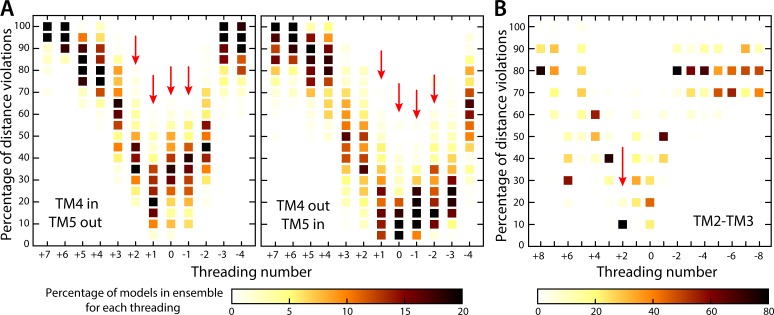
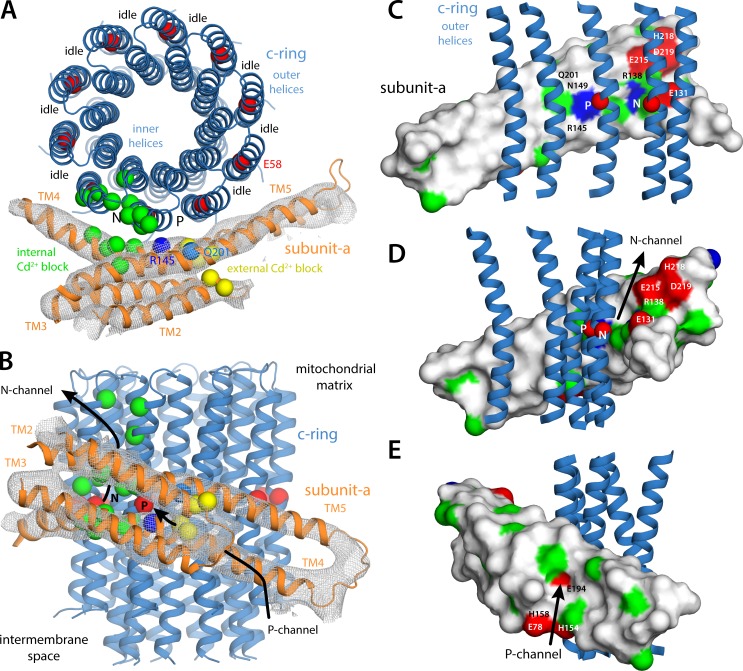
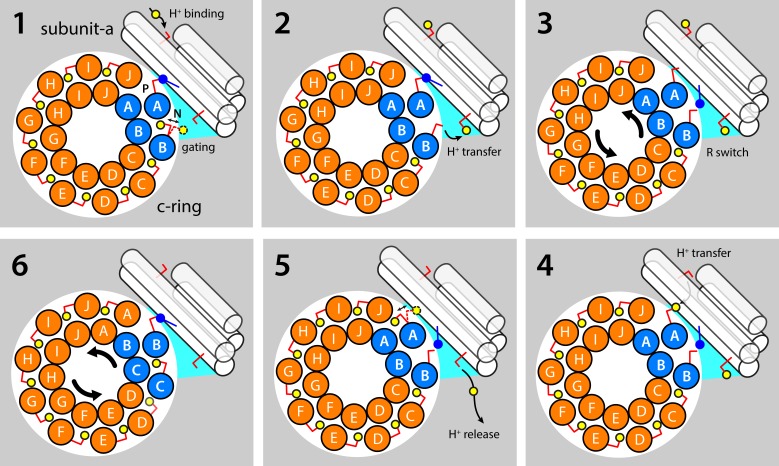
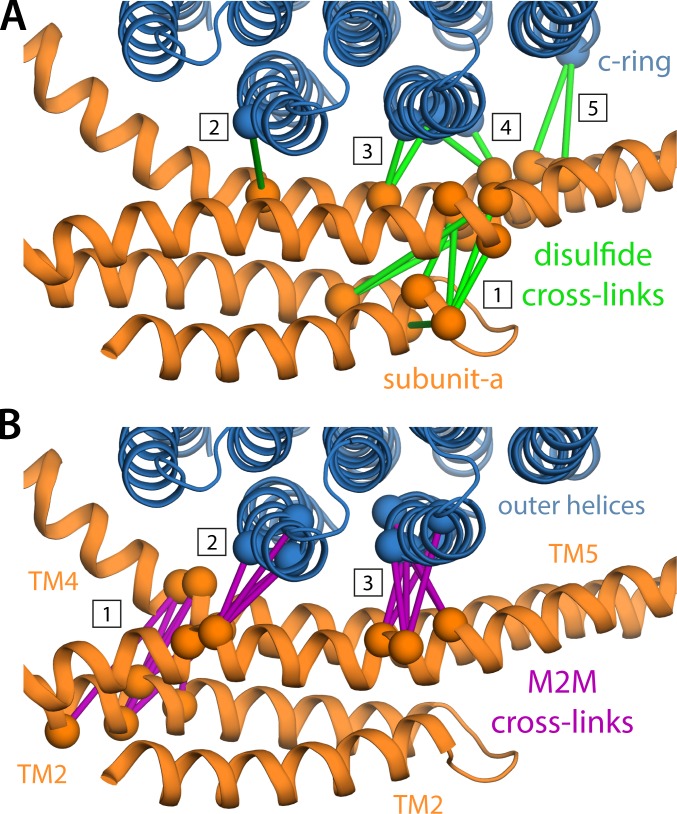
Two subunits within the transmembrane domain of the ATP synthase-the c-ring and subunit a-energize the production of 90% of cellular ATP by transducing an electrochemical gradient of H or Na into rotational motion. The nature of this turbine-like energy conversion mechanism has been elusive for decades, owing to the lack of definitive structural information on subunit a or its c-ring interface. In a recent breakthrough, several structures of this complex were resolved by cryo-electron microscopy (cryo-EM), but the modest resolution of the data has led to divergent interpretations. Moreover, the unexpected architecture of the complex has cast doubts on a wealth of earlier biochemical analyses conducted to probe this structure. Here, we use quantitative molecular-modeling methods to derive a structure of the a-c complex that is not only objectively consistent with the cryo-EM data, but also with correlated mutation analyses of both subunits and with prior cross-linking and cysteine accessibility measurements. This systematic, integrative approach reveals unambiguously the topology of subunit a and its relationship with the c-ring. Mapping of known Cd block sites and conserved protonatable residues onto the structure delineates two noncontiguous pathways across the complex, connecting two adjacent proton-binding sites in the c-ring to the space on either side of the membrane. The location of these binding sites and of a strictly conserved arginine on subunit a, which serves to prevent protons from hopping between them, explains the directionality of the rotary mechanism and its strict coupling to the proton-motive force. Additionally, mapping of mutations conferring resistance to oligomycin unexpectedly reveals that this prototypical inhibitor may bind to two distinct sites at the a-c interface, explaining its ability to block the mechanism of the enzyme irrespective of the direction of rotation of the c-ring. In summary, this study is a stepping stone toward establishing the mechanism of the ATP synthase at the atomic level.
ATP合酶跨膜结构域中的两个亚基——c环和亚基a——通过将H或Na的电化学梯度转化为旋转运动,为细胞中90%的ATP生成提供能量。由于缺乏关于亚基a或其与c环界面的确切结构信息,这种类似涡轮机的能量转换机制的本质几十年来一直难以捉摸。最近有一项突破,通过冷冻电子显微镜(cryo-EM)解析了该复合物的几种结构,但数据的中等分辨率导致了不同的解释。此外,该复合物出人意料的结构对之前为探究其结构而进行的大量生化分析产生了质疑。在这里,我们使用定量分子建模方法得出a-c复合物的结构,该结构不仅客观上与冷冻电子显微镜数据一致,而且与两个亚基的相关突变分析以及先前的交联和半胱氨酸可及性测量结果一致。这种系统的、综合的方法明确揭示了亚基a的拓扑结构及其与c环的关系。将已知的镉阻断位点和保守的可质子化残基映射到该结构上,描绘了穿过复合物的两条不连续途径,将c环中两个相邻的质子结合位点与膜两侧的空间连接起来。这些结合位点的位置以及亚基a上一个严格保守的精氨酸的位置,可防止质子在它们之间跳跃,这解释了旋转机制的方向性及其与质子动力的严格耦合。此外,对赋予寡霉素抗性的突变进行映射,意外地发现这种典型抑制剂可能在a-c界面结合到两个不同的位点,这解释了它无论c环旋转方向如何都能阻断酶机制的能力。总之,这项研究是朝着在原子水平上建立ATP合酶机制迈出的一块垫脚石。