Riva Beatrice, De Dominici Marco, Gnemmi Ilaria, Mariani Samanta A, Minassi Alberto, Minieri Valentina, Salomoni Paolo, Canonico Pier Luigi, Genazzani Armando A, Calabretta Bruno, Condorelli Fabrizio
Department of Pharmacological Sciences, Università del Piemonte Orientale "A. Avogadro", 28100 Novara, Italy.
Sidney Kimmel Cancer Center, Thomas Jefferson University, Philadelphia 19107, PA, USA.
Oncotarget. 2016 Dec 6;7(49):81555-81570. doi: 10.18632/oncotarget.13146.
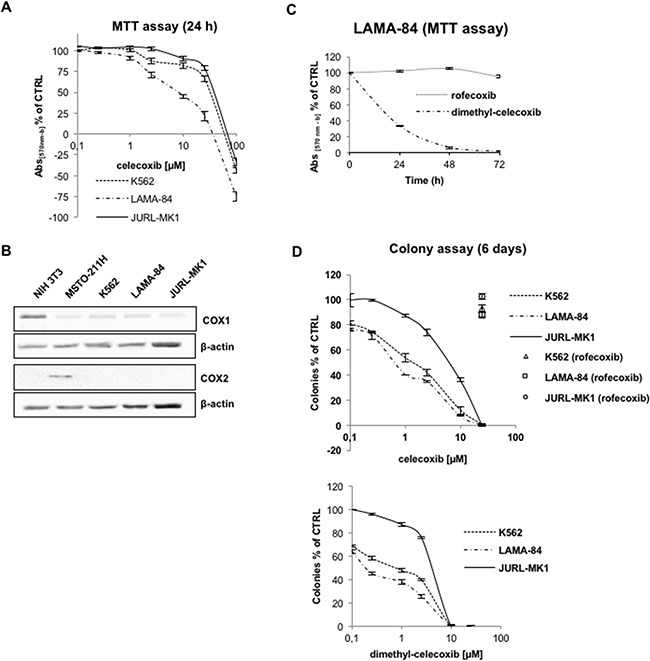
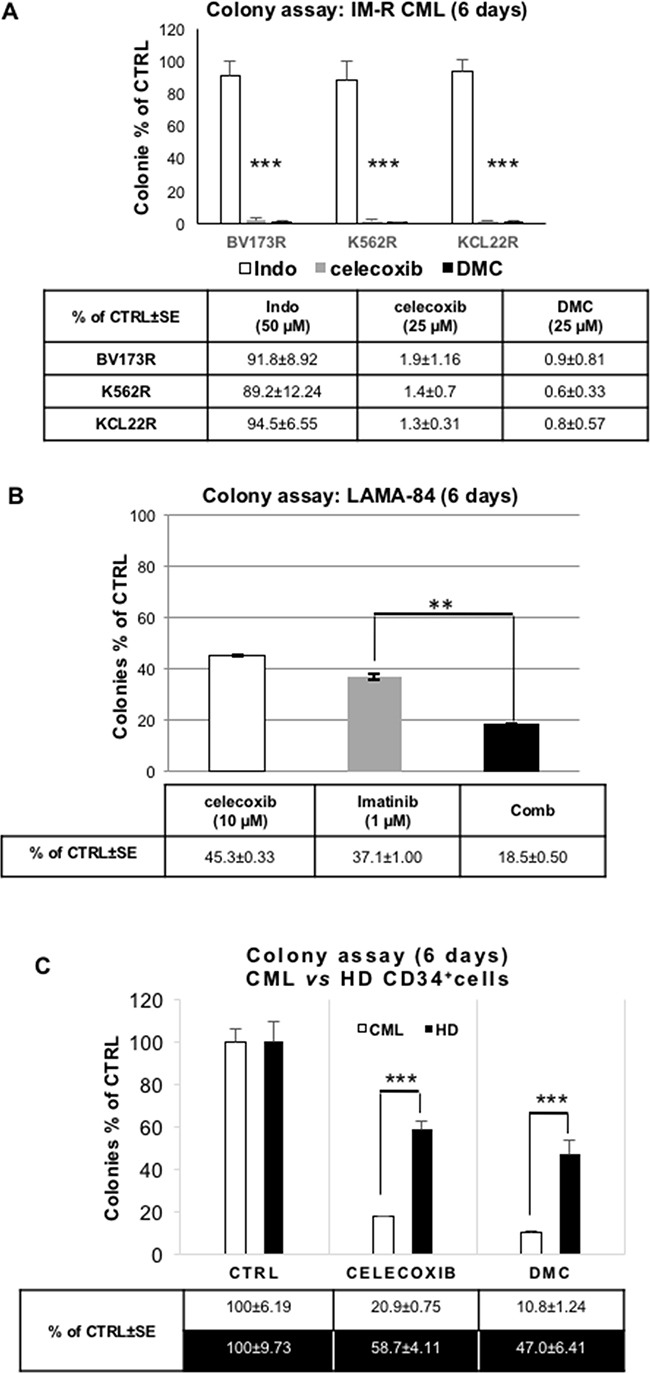
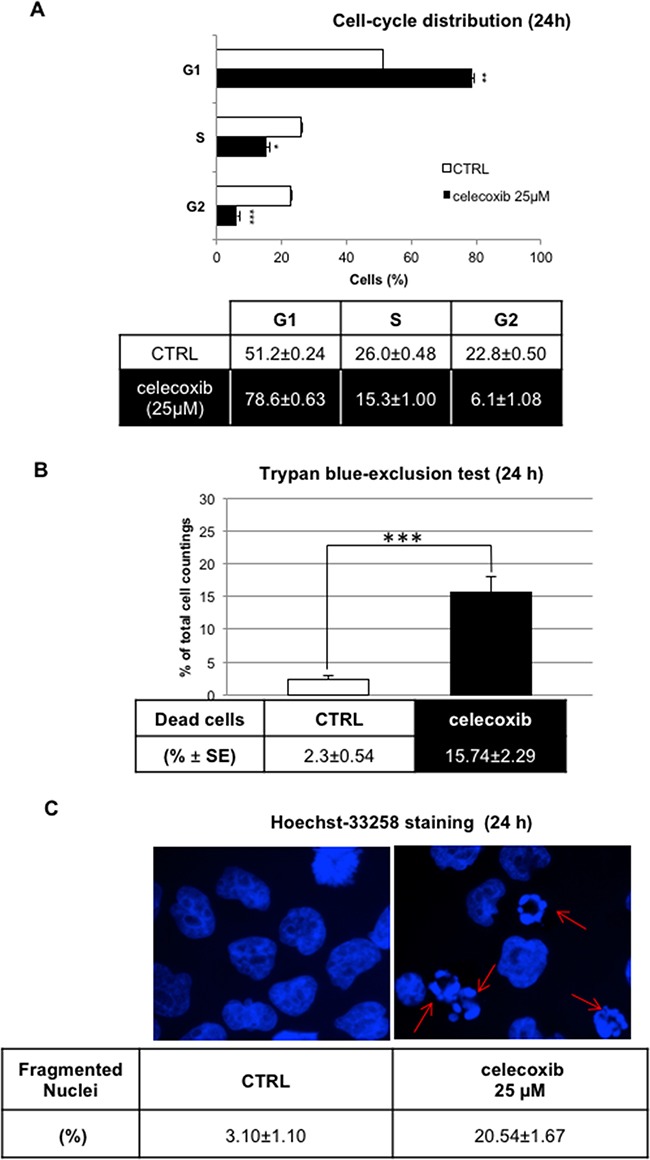
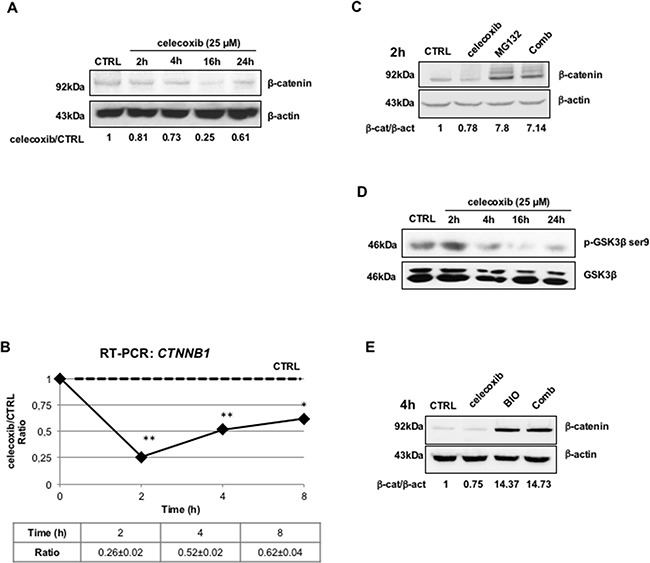
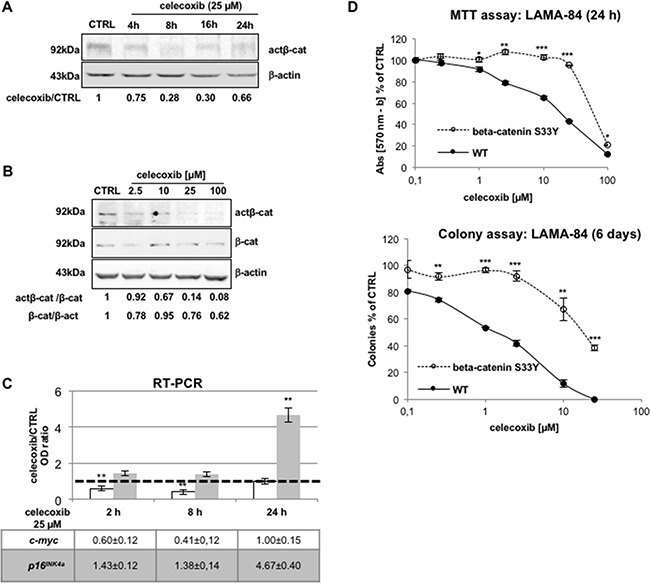
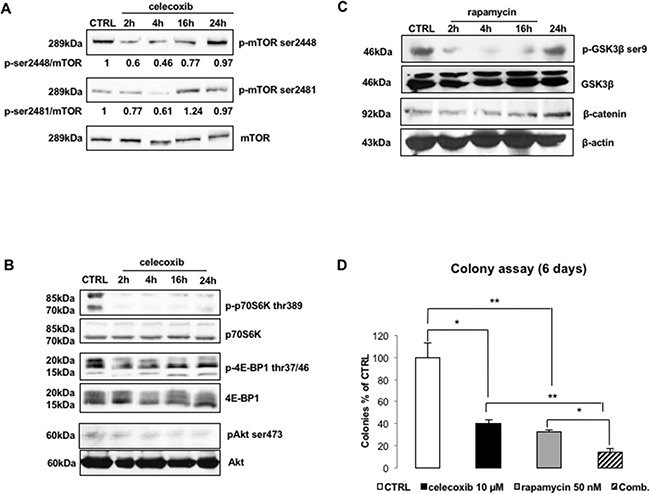
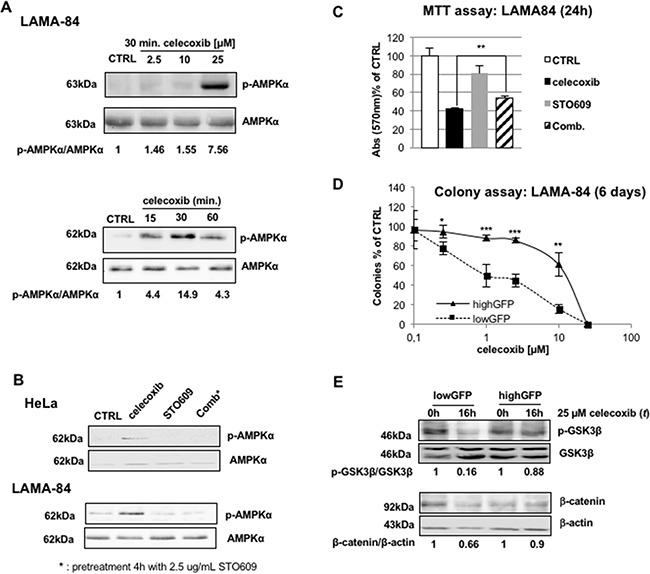
CML is effectively treated with tyrosine kinase inhibitors (TKIs). However, the efficacy of these drugs is confined to the chronic phase of the disease and development of resistance to TKIs remains a pressing issue. The anti-inflammatory COX2 inhibitor celecoxib has been utilized as anti-tumour drug due to its anti-proliferative activity. However, its effects in hematological malignancies, in particular CML, have not been investigated yet. Thus, we tested biological effects and mechanisms of action of celecoxib in Philadelphia-positive (Ph+) CML and ALL cells.We show here that celecoxib suppresses the growth of Ph+ cell lines by increasing G1-phase and apoptotic cells and reducing S- and G2-phase cells. These effects were independent of COX2 inhibition but required the rapid activation of AMP-activated protein kinase (AMPK) and the consequent inhibition mTORC1 and 2. Treatment with celecoxib also restored GSK3β function and led to down-regulation of β-catenin activity through transcriptional and post-translational mechanisms, two effects likely to contribute to Ph+ cell growth suppression by celecoxib.Celecoxib inhibited colony formation of TKI-resistant Ph+ cell lines including those with the T315I BCR-ABL mutation and acted synergistically with imatinib in suppressing colony formation of TKI-sensitive Ph+ cell lines. Finally, it suppressed colony formation of CD34+ cells from CML patients, while sparing most CD34+ progenitors from healthy donors, and induced apoptosis of primary Ph+ ALL cells.Together, these findings indicate that celecoxib may serve as a COX2-independent lead compound to simultaneously target the mTOR and β-catenin pathways, key players in the resistance of CML stem cells to TKIs.
慢性粒细胞白血病(CML)可通过酪氨酸激酶抑制剂(TKIs)得到有效治疗。然而,这些药物的疗效仅限于疾病的慢性期,对TKIs产生耐药性仍是一个紧迫的问题。抗炎性环氧化酶2(COX2)抑制剂塞来昔布因其抗增殖活性已被用作抗肿瘤药物。然而,其在血液系统恶性肿瘤,特别是CML中的作用尚未得到研究。因此,我们测试了塞来昔布在费城染色体阳性(Ph+)CML和急性淋巴细胞白血病(ALL)细胞中的生物学效应及作用机制。我们在此表明,塞来昔布通过增加G1期细胞和凋亡细胞以及减少S期和G2期细胞来抑制Ph+细胞系的生长。这些效应与COX2抑制无关,但需要快速激活AMP激活的蛋白激酶(AMPK)以及随后抑制哺乳动物雷帕霉素靶蛋白复合物1和2(mTORC1和2)。用塞来昔布治疗还恢复了糖原合成酶激酶3β(GSK3β)的功能,并通过转录和翻译后机制导致β-连环蛋白活性下调,这两种效应可能有助于塞来昔布抑制Ph+细胞生长。塞来昔布抑制了对TKI耐药的Ph+细胞系的集落形成,包括那些具有T315I BCR-ABL突变的细胞系,并且在抑制对TKI敏感的Ph+细胞系的集落形成方面与伊马替尼协同作用。最后,它抑制了CML患者CD34+细胞的集落形成,同时使大多数健康供体的CD34+祖细胞免受影响,并诱导原发性Ph+ ALL细胞凋亡。总之,这些发现表明塞来昔布可能作为一种不依赖COX2的先导化合物,同时靶向mTOR和β-连环蛋白途径,这两条途径是CML干细胞对TKIs耐药的关键因素。