Cacheux Marine, Blum Ariane, Sébastien Muriel, Wozny Anne Sophie, Brocard Julie, Mamchaoui Kamel, Mouly Vincent, Roux-Buisson Nathalie, Rendu John, Monnier Nicole, Krivosic Renée, Allen Paul, Lacour Arnaud, Lunardi Joël, Fauré Julien, Marty Isabelle
INSERM U836, Grenoble Institut des Neurosciences, Equipe Muscle et Pathologies, Grenoble, France.
Université Joseph Fourier, Grenoble, France.
J Neuromuscul Dis. 2015 Nov 20;2(4):421-432. doi: 10.3233/JND-150073.
Central Core Disease (CCD) is a congenital myopathy often resulting from a mutation in RYR1 gene. Mutations in RyR1 can increase or decrease channel activity, or induce a reduction in the amount of protein. The consequences of a single mutation are sometimes multiple and the analysis of the functional effects is complex.
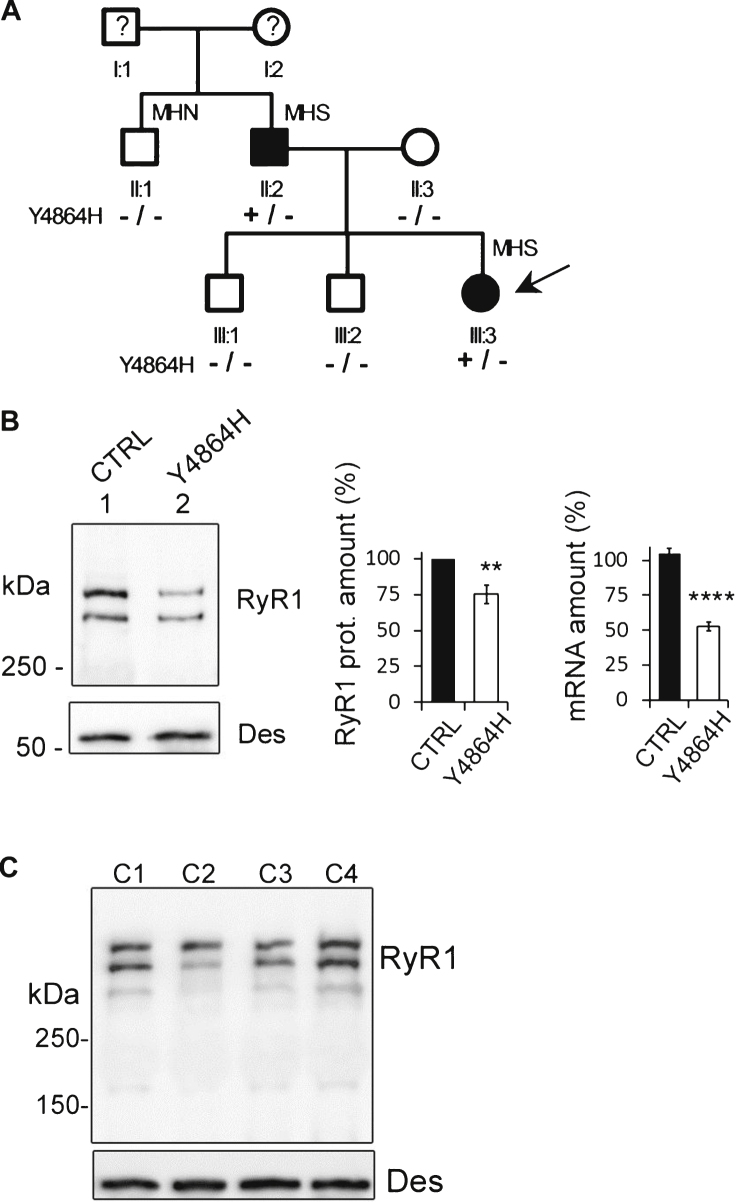
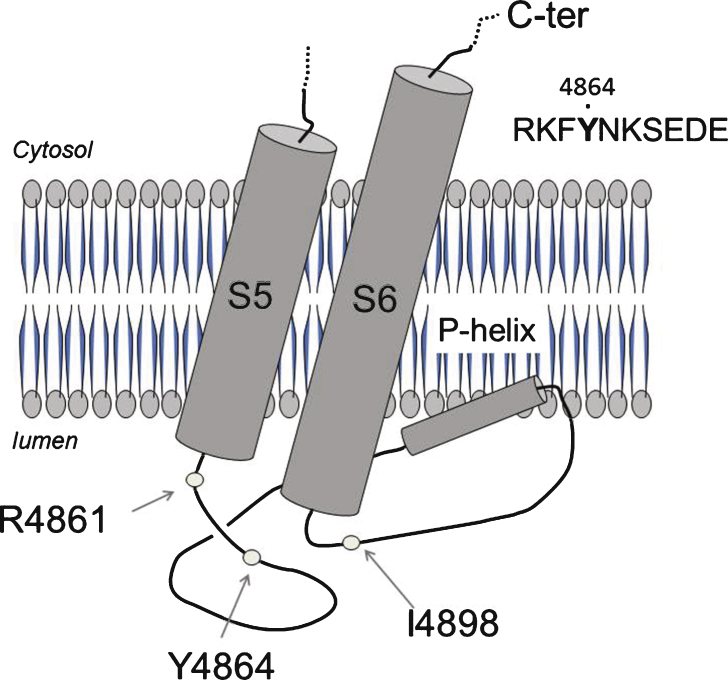
The consequences of the p.Y4864H mutation identified in a CCD patient have been studied regarding both RyR1 function and amount.
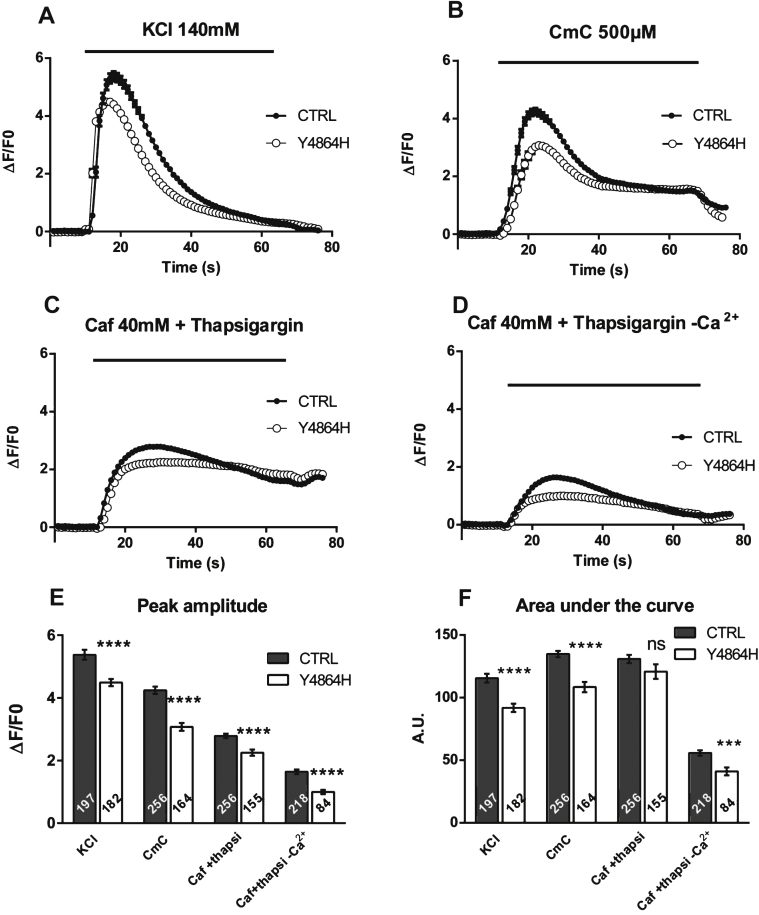
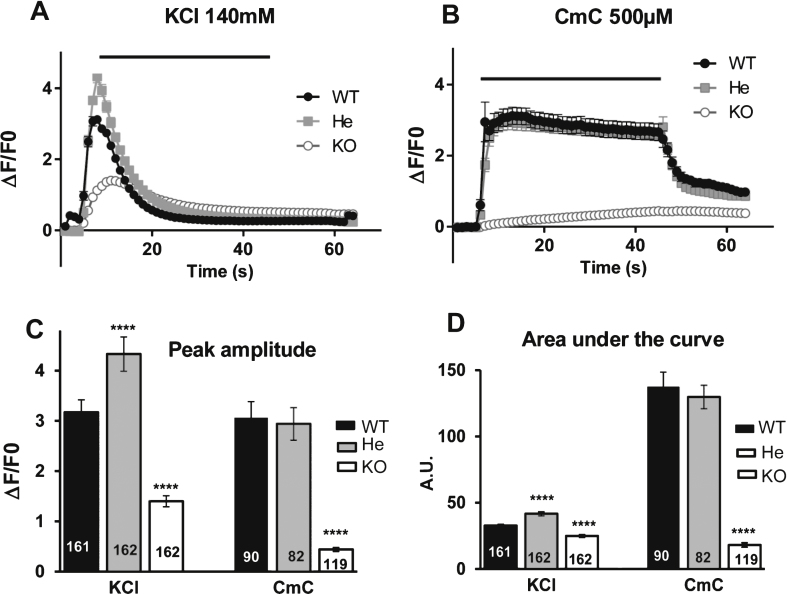
The amount of RyR1 in human and mouse muscles was evaluated using qRT-PCR and quantitative Western blot, and calcium release was studied using calcium imaging on primary cultures. The results were compared between human and mouse.
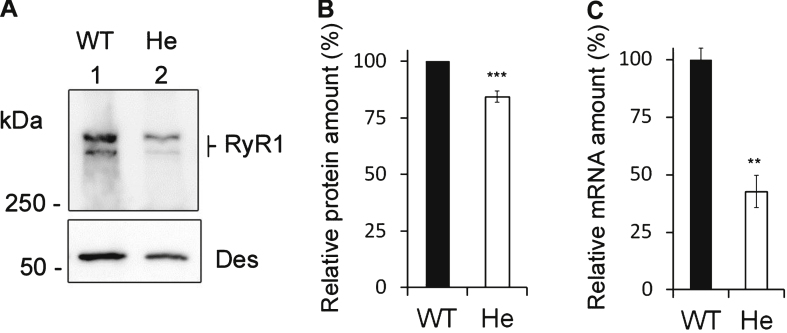
The p.Y4864H mutation induced an alteration of calcium release, and in addition was associated to a reduction in the amount of RyR1 in the patient's muscle. This suggests two possible pathophysiological mechanisms: the alteration of calcium release could result from a modification of the channel properties of RyR1 or from a RyR1 reduction. In order to discriminate between the two hypotheses, we used the heterozygous RyR1 knockout (RyR1+/-) mouse model showing a comparable RyR1 protein reduction. No reduction in calcium release was observed in primary muscle culture from these mice, and no muscle weakness was measured.
Because the reduction in the amount of RyR1 protein has no functional consequences in the murine model, the muscle weakness observed in the patient is most likely the result of a modification of the calcium channel function of RyR1 due to the p.Y4864H mutation.
中央轴空病(CCD)是一种先天性肌病,通常由RYR1基因突变引起。RyR1基因突变可增加或降低通道活性,或导致蛋白质数量减少。单个突变的后果有时是多方面的,功能效应分析较为复杂。
研究在一名中央轴空病患者中鉴定出的p.Y4864H突变对RyR1功能和数量的影响。
使用qRT-PCR和定量蛋白质免疫印迹法评估人和小鼠肌肉中RyR1的数量,并在原代培养物中通过钙成像研究钙释放情况。对人和小鼠之间的结果进行比较。
p.Y4864H突变导致钙释放改变,此外还与患者肌肉中RyR1数量减少有关。这提示了两种可能的病理生理机制:钙释放改变可能是由于RyR1通道特性改变或RyR1减少所致。为了区分这两种假设,我们使用了杂合型RyR1基因敲除(RyR1+/-)小鼠模型,该模型显示出类似的RyR1蛋白减少。在这些小鼠的原代肌肉培养物中未观察到钙释放减少,也未检测到肌肉无力。
由于在小鼠模型中RyR1蛋白数量减少没有功能后果,因此患者中观察到的肌肉无力很可能是由于p.Y4864H突变导致RyR1钙通道功能改变的结果。