Ambrosini Giovanna, Dreos René, Kumar Sunil, Bucher Philipp
School of Life Sciences, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015, Lausanne, Switzerland.
Swiss Institute of Bioinformatics (SIB), CH-1015, Lausanne, Switzerland.
BMC Genomics. 2016 Nov 18;17(1):938. doi: 10.1186/s12864-016-3288-8.
ChIP-seq and related high-throughput chromatin profilig assays generate ever increasing volumes of highly valuable biological data. To make sense out of it, biologists need versatile, efficient and user-friendly tools for access, visualization and itegrative analysis of such data.
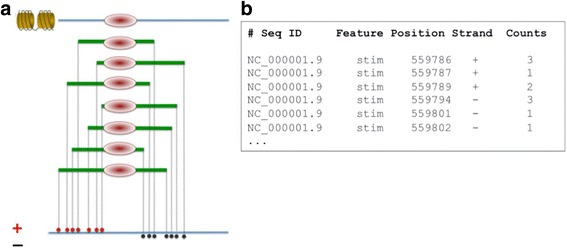
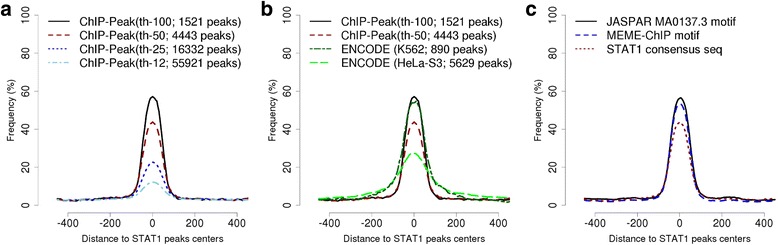
Here we present the ChIP-Seq command line tools and web server, implementing basic algorithms for ChIP-seq data analysis starting with a read alignment file. The tools are optimized for memory-efficiency and speed thus allowing for processing of large data volumes on inexpensive hardware. The web interface provides access to a large database of public data. The ChIP-Seq tools have a modular and interoperable design in that the output from one application can serve as input to another one. Complex and innovative tasks can thus be achieved by running several tools in a cascade.
The various ChIP-Seq command line tools and web services either complement or compare favorably to related bioinformatics resources in terms of computational efficiency, ease of access to public data and interoperability with other web-based tools. The ChIP-Seq server is accessible at http://ccg.vital-it.ch/chipseq/ .
染色质免疫沉淀测序(ChIP-seq)及相关的高通量染色质分析方法产生了越来越多极具价值的生物学数据。为了理解这些数据,生物学家需要通用、高效且用户友好的工具来访问、可视化和综合分析此类数据。
在此,我们展示了ChIP-Seq命令行工具和网络服务器,它们实现了从读取比对文件开始的ChIP-seq数据分析的基本算法。这些工具针对内存效率和速度进行了优化,从而能够在低成本硬件上处理大量数据。网络界面提供了对大型公共数据库的访问。ChIP-Seq工具具有模块化和可互操作的设计,即一个应用程序的输出可以作为另一个应用程序的输入。因此,通过级联运行多个工具可以完成复杂和创新的任务。
各种ChIP-Seq命令行工具和网络服务在计算效率、对公共数据的访问便利性以及与其他基于网络的工具的互操作性方面,要么相互补充,要么优于相关的生物信息学资源。ChIP-Seq服务器可通过http://ccg.vital-it.ch/chipseq/访问。