Takahashi Hisashi, Noto Yu-Ichi, Makita Naoki, Kushimura-Okada Yukie, Ishii Ryotaro, Tanaka Akihiro, Ohara Tomoyuki, Nakane Shunya, Higuchi Osamu, Nakagawa Masanori, Mizuno Toshiki
Department of Neurology, Kyoto Prefectural University of Medicine Graduate School of Medical Science, 465 Kajii-cho, Kamigyo-ku, Kyoto, 602-0841, Japan.
Department of Neurology, National Hospital Organization Maizuru Medical Center, Maizuru, Japan.
BMC Neurol. 2016 Nov 18;16(1):229. doi: 10.1186/s12883-016-0758-1.
Myasthenic symptoms can be present in patients with amyotrophic lateral sclerosis (ALS). These symptoms have been considered to be caused by the degeneration of distal motor neurons and the neuromuscular junction (NMJ). Recent studies suggested that antibody to low-density lipoprotein receptor-related protein 4 (LRP4) was a pathogenic agent of myasthenia gravis (MG), and it was also detected in ALS patients.
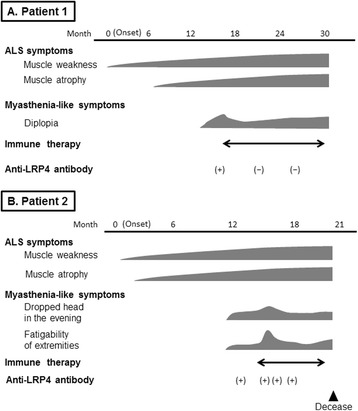
Patient 1: A 58-year-old Japanese man developed progressive weakness and subsequent myasthenic symptoms including oculomotor disturbance. Clinical examination and electrophysiological studies confirmed upper and lower motor neuron involvement and NMJ dysfunction, and anti-LRP4 antibody was detected in his serum. A series of immunotherapies, including steroid pulse therapy, intravenous immunoglobulin, and plasmapheresis, was performed, and the myasthenic symptoms partially improved. The titer of anti-LRP4 antibody subsequently decreased. However, the therapeutic effect was transient, and ALS symptoms progressed. His clinical findings fulfilled the criteria of probable ALS using the Awaji criteria. Patient 2: A 74-year-old Japanese man suffered from progressive weakness of all limbs and dropped head in the evening. He complained of diplopia with a lateral horizontal gaze. Probable ALS was diagnosed because of the upper and lower motor neuron signs, whereas anti-LRP4 antibody was detected. Several immunotherapies were administered, and the myasthenic symptoms partially responded to each therapy. However, the truncal muscle weakness progressed, and he died of respiratory failure.
We report two anti-LRP4 antibody-seropositive ALS patients with myasthenia who were not typical of ALS patients, and showed partial responses to immunotherapies. The anti-LRP4 antibody-seropositive status may influence developing ALS and cause additional ALS symptoms.
肌萎缩侧索硬化症(ALS)患者可能出现肌无力症状。这些症状被认为是由远端运动神经元和神经肌肉接头(NMJ)的退化引起的。最近的研究表明,低密度脂蛋白受体相关蛋白4(LRP4)抗体是重症肌无力(MG)的致病因子,并且在ALS患者中也检测到了该抗体。
病例1:一名58岁的日本男性出现进行性肌无力,随后出现包括动眼障碍在内的肌无力症状。临床检查和电生理研究证实存在上下运动神经元受累及NMJ功能障碍,其血清中检测到抗LRP4抗体。进行了一系列免疫治疗,包括类固醇冲击疗法、静脉注射免疫球蛋白和血浆置换,肌无力症状部分改善。抗LRP4抗体滴度随后下降。然而,治疗效果是短暂的,ALS症状仍在进展。根据阿波岐标准,他的临床症状符合可能的ALS标准。病例2:一名74岁的日本男性四肢进行性无力,傍晚时出现垂头症状。他主诉向外侧水平注视时出现复视。由于存在上下运动神经元体征,诊断为可能的ALS,同时检测到抗LRP4抗体。给予了几种免疫治疗,肌无力症状对每种治疗都有部分反应。然而,躯干肌无力进展,他死于呼吸衰竭。
我们报告了两名抗LRP4抗体血清阳性的ALS合并肌无力患者,他们并非典型的ALS患者,且对免疫治疗有部分反应。抗LRP4抗体血清阳性状态可能影响ALS的发展并导致额外的ALS症状。