Yoo Yun Joo, Sun Lei, Poirier Julia G, Paterson Andrew D, Bull Shelley B
Department of Mathematics Education, Seoul National University, Seoul, South Korea.
Interdisciplinary Program in Bioinformatics, Seoul National University, Seoul, South Korea.
Genet Epidemiol. 2017 Feb;41(2):108-121. doi: 10.1002/gepi.22024. Epub 2016 Nov 25.
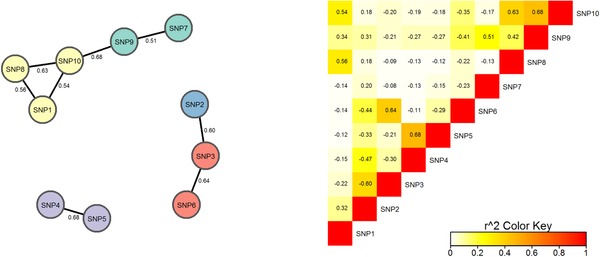
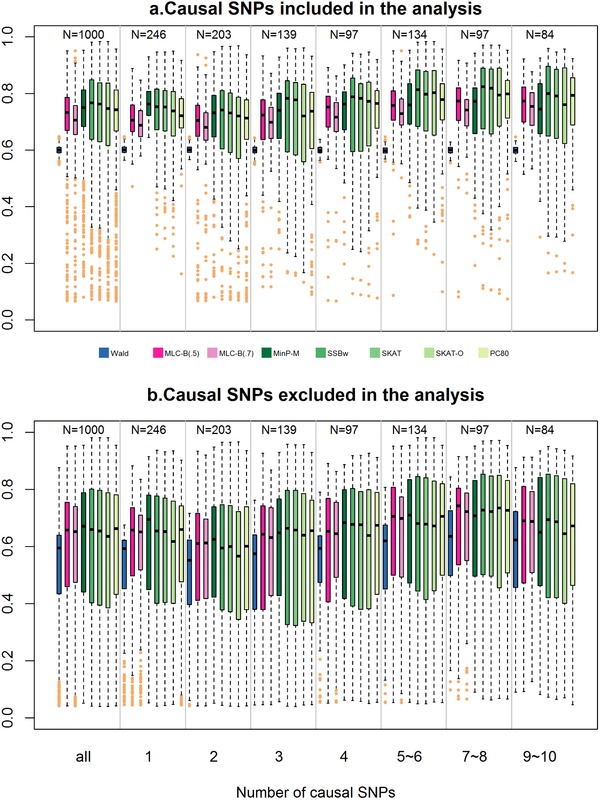
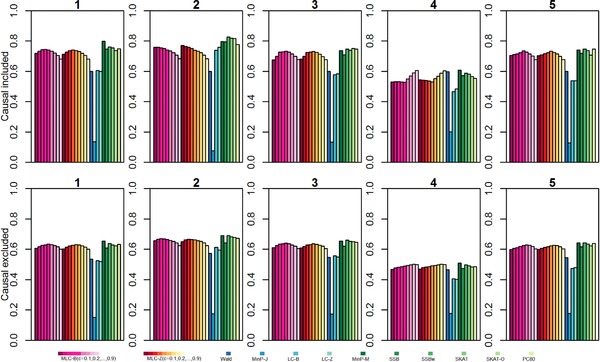
By jointly analyzing multiple variants within a gene, instead of one at a time, gene-based multiple regression can improve power, robustness, and interpretation in genetic association analysis. We investigate multiple linear combination (MLC) test statistics for analysis of common variants under realistic trait models with linkage disequilibrium (LD) based on HapMap Asian haplotypes. MLC is a directional test that exploits LD structure in a gene to construct clusters of closely correlated variants recoded such that the majority of pairwise correlations are positive. It combines variant effects within the same cluster linearly, and aggregates cluster-specific effects in a quadratic sum of squares and cross-products, producing a test statistic with reduced degrees of freedom (df) equal to the number of clusters. By simulation studies of 1000 genes from across the genome, we demonstrate that MLC is a well-powered and robust choice among existing methods across a broad range of gene structures. Compared to minimum P-value, variance-component, and principal-component methods, the mean power of MLC is never much lower than that of other methods, and can be higher, particularly with multiple causal variants. Moreover, the variation in gene-specific MLC test size and power across 1000 genes is less than that of other methods, suggesting it is a complementary approach for discovery in genome-wide analysis. The cluster construction of the MLC test statistics helps reveal within-gene LD structure, allowing interpretation of clustered variants as haplotypic effects, while multiple regression helps to distinguish direct and indirect associations.
通过同时分析一个基因内的多个变异而非逐个分析,基于基因的多重回归可以提高遗传关联分析的效能、稳健性及可解释性。我们基于HapMap亚洲单倍型,在具有连锁不平衡(LD)的现实性状模型下,研究用于常见变异分析的多重线性组合(MLC)检验统计量。MLC是一种定向检验,它利用基因中的LD结构构建紧密相关变异的簇,这些变异被重新编码,使得大多数成对相关性为正。它线性组合同一簇内的变异效应,并在平方和与交叉积的二次和中汇总特定簇的效应,产生一个自由度(df)降低至簇数量的检验统计量。通过对全基因组1000个基因的模拟研究,我们证明,在广泛的基因结构范围内,MLC在现有方法中是一种效能良好且稳健的选择。与最小P值、方差成分和主成分方法相比,MLC的平均效能从不比其他方法低很多,并且可能更高,特别是存在多个因果变异时。此外,1000个基因中特定基因的MLC检验规模和效能的变化小于其他方法,这表明它是全基因组分析中一种互补的发现方法。MLC检验统计量的簇构建有助于揭示基因内的LD结构,允许将成簇变异解释为单倍型效应,而多重回归有助于区分直接和间接关联。