Lun Aaron T L, McCarthy Davis J, Marioni John C
Cancer Research UK Cambridge Institute, Cambridge, UK.
EMBL European Bioinformatics Institute, Cambridge, UK; St Vincent's Institute of Medical Research, Fitzroy, Australia.
F1000Res. 2016 Aug 31;5:2122. doi: 10.12688/f1000research.9501.2. eCollection 2016.
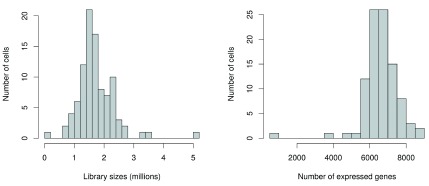
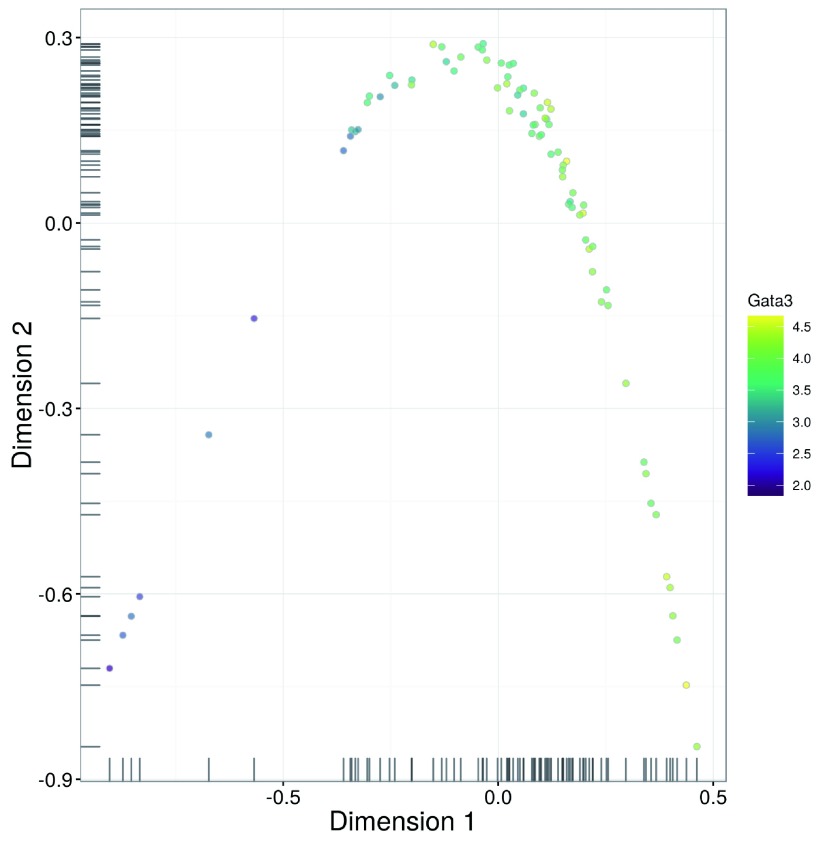
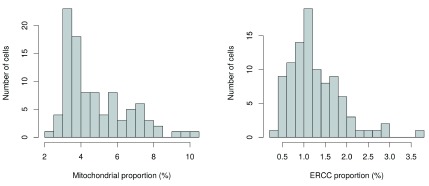
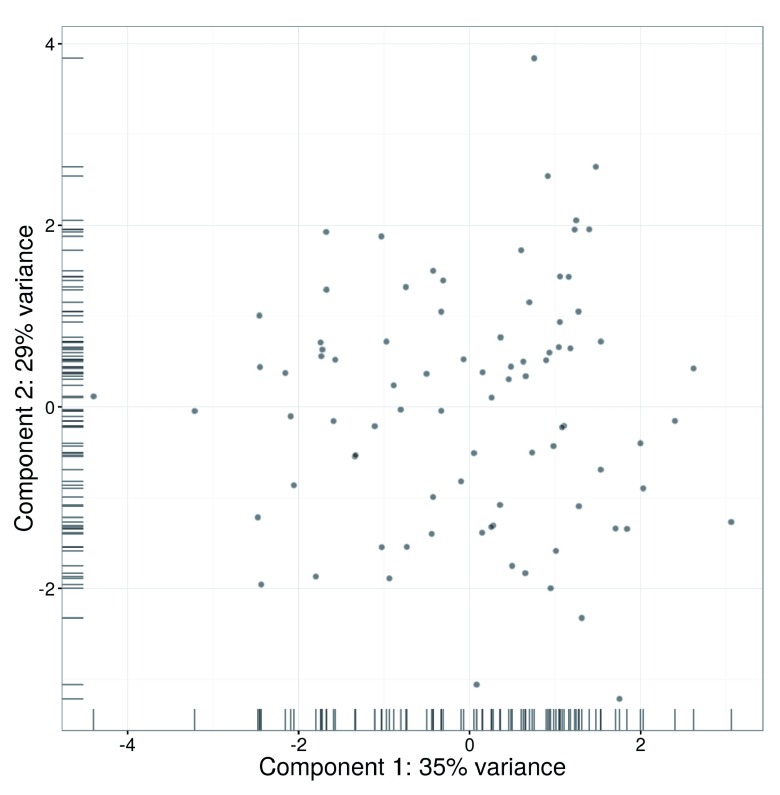
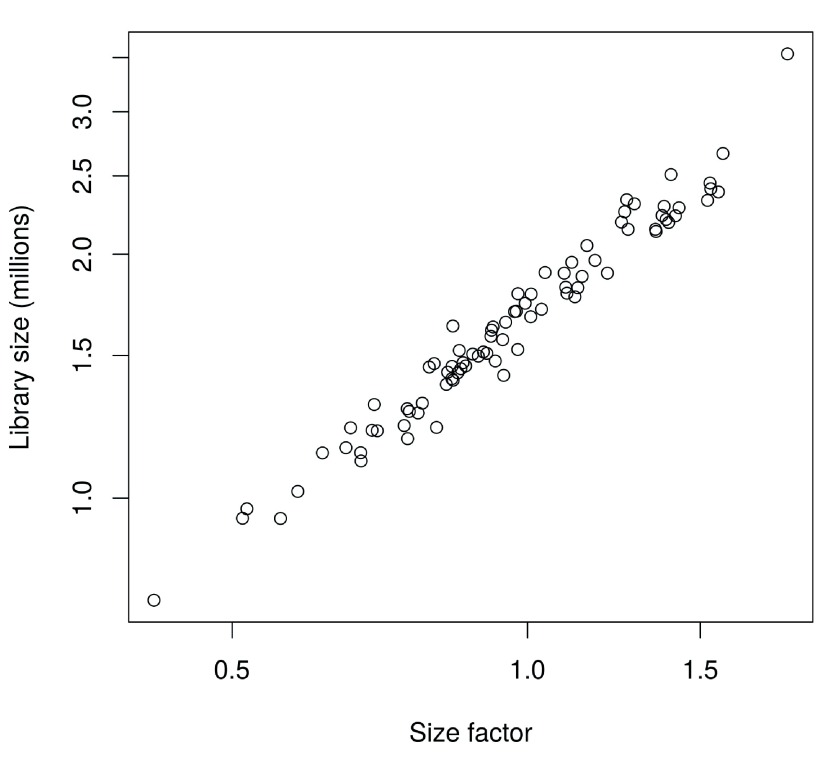
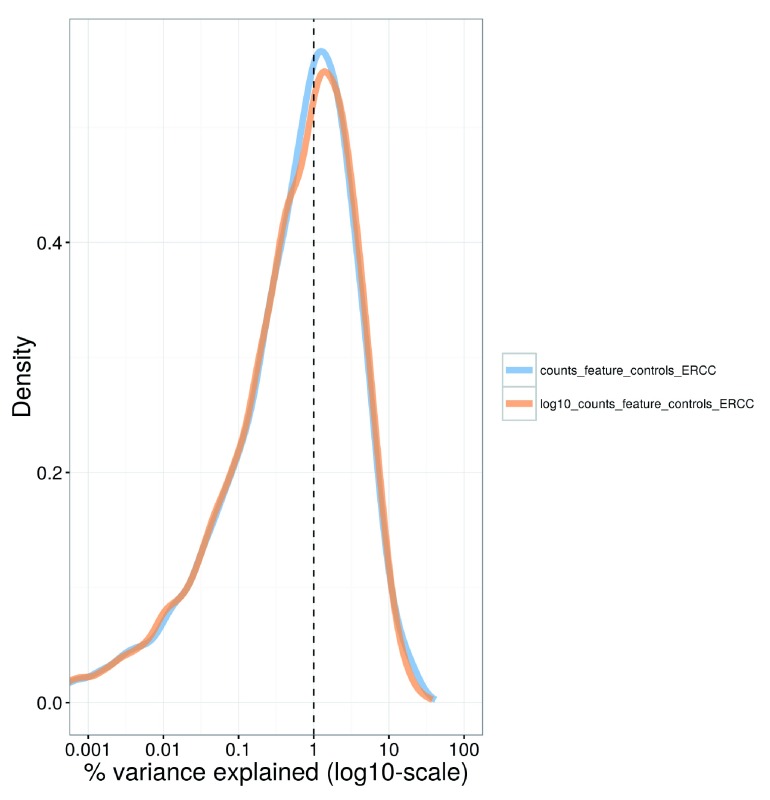
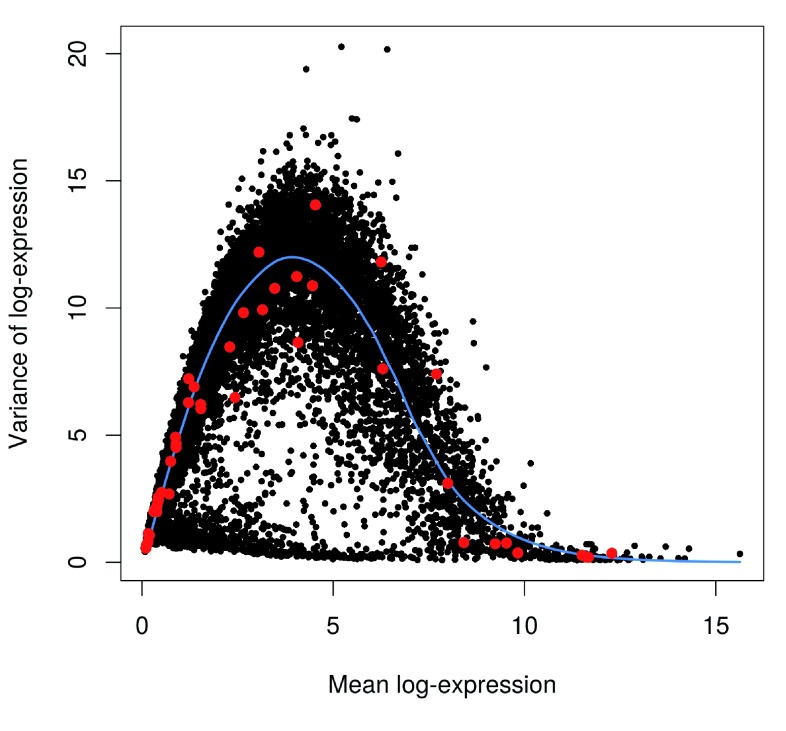
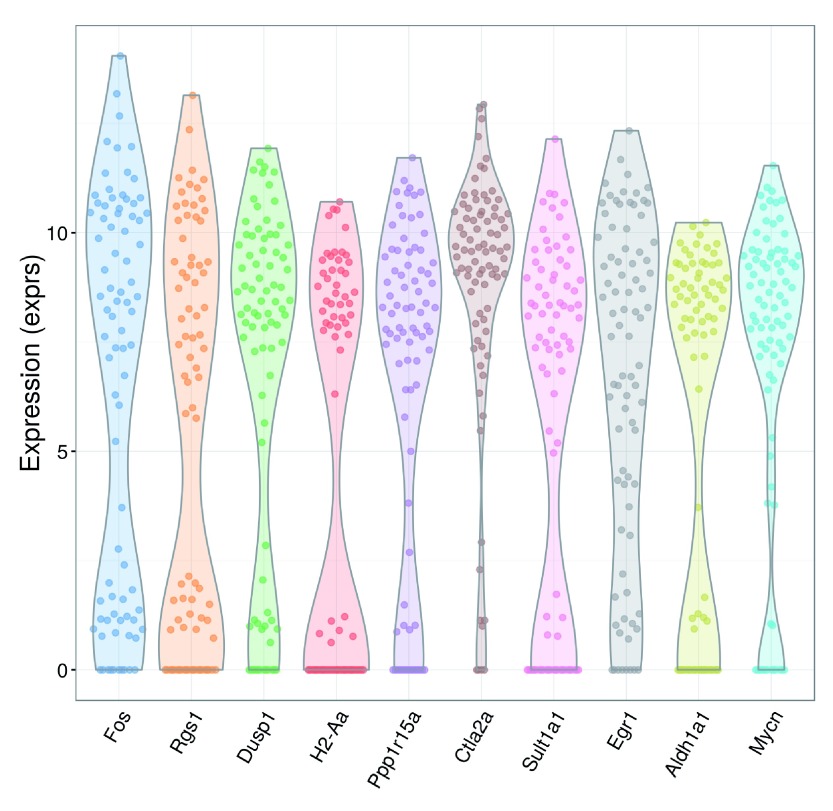
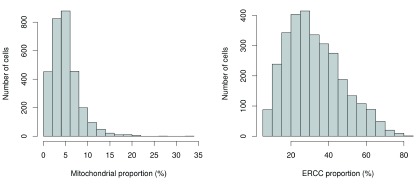
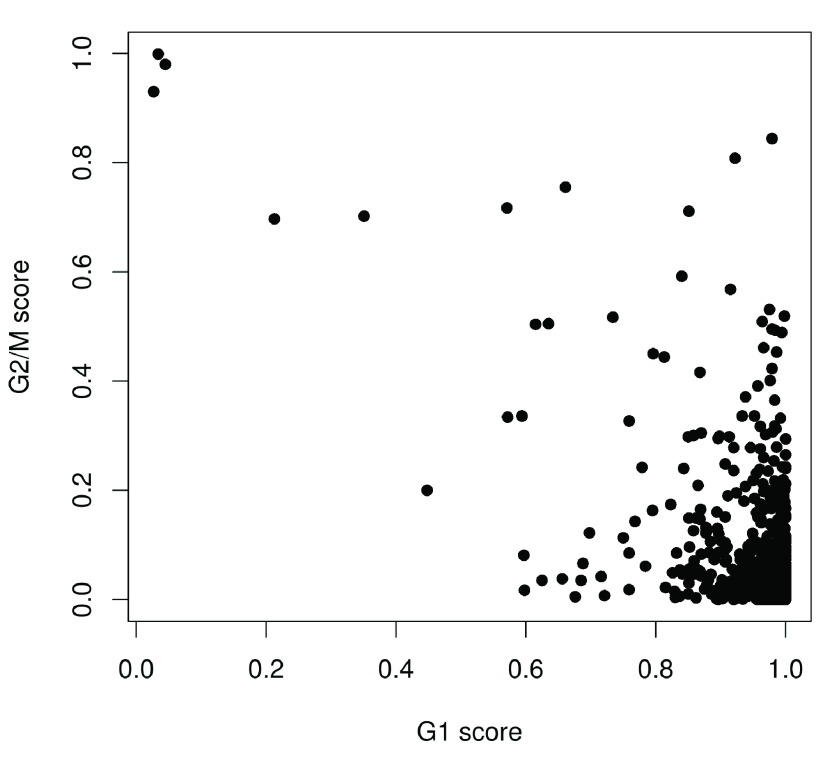
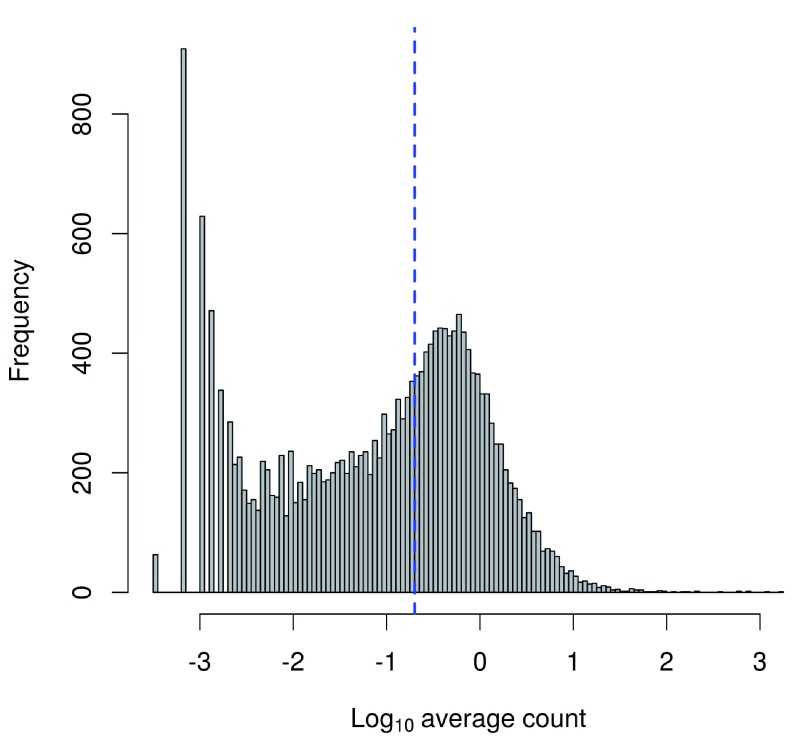
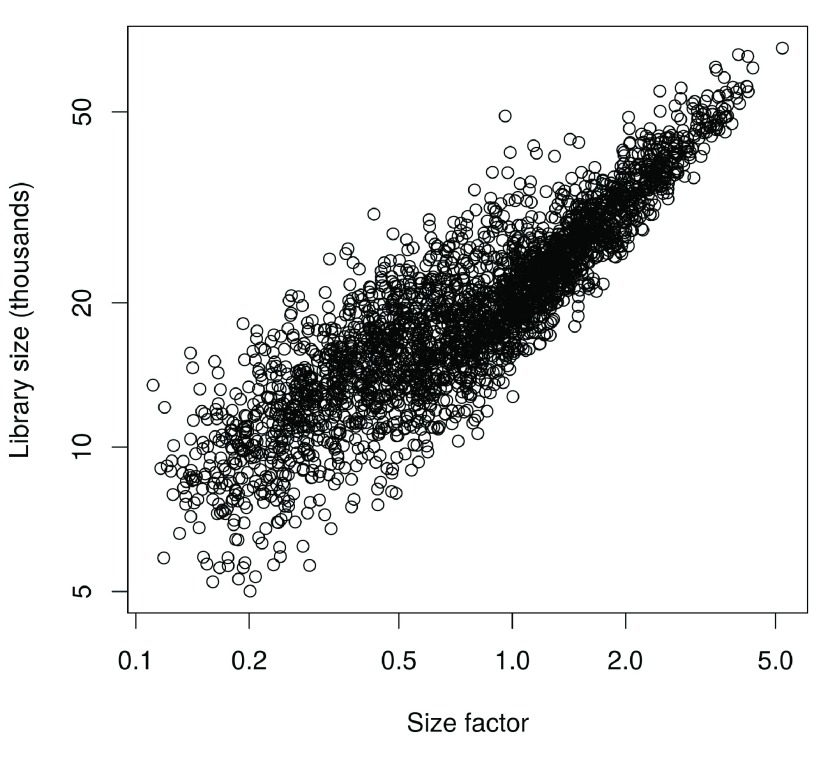
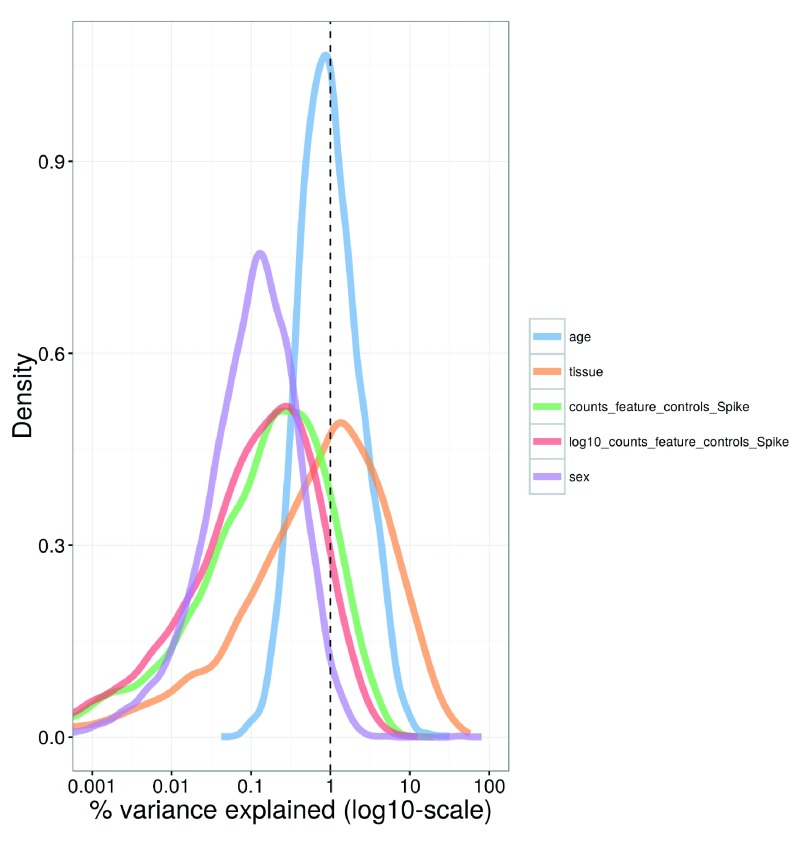
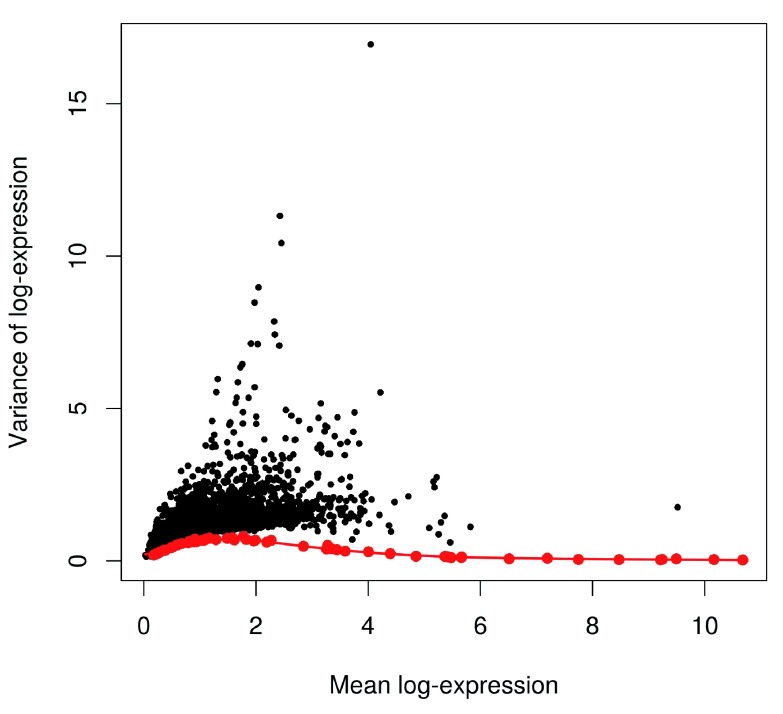
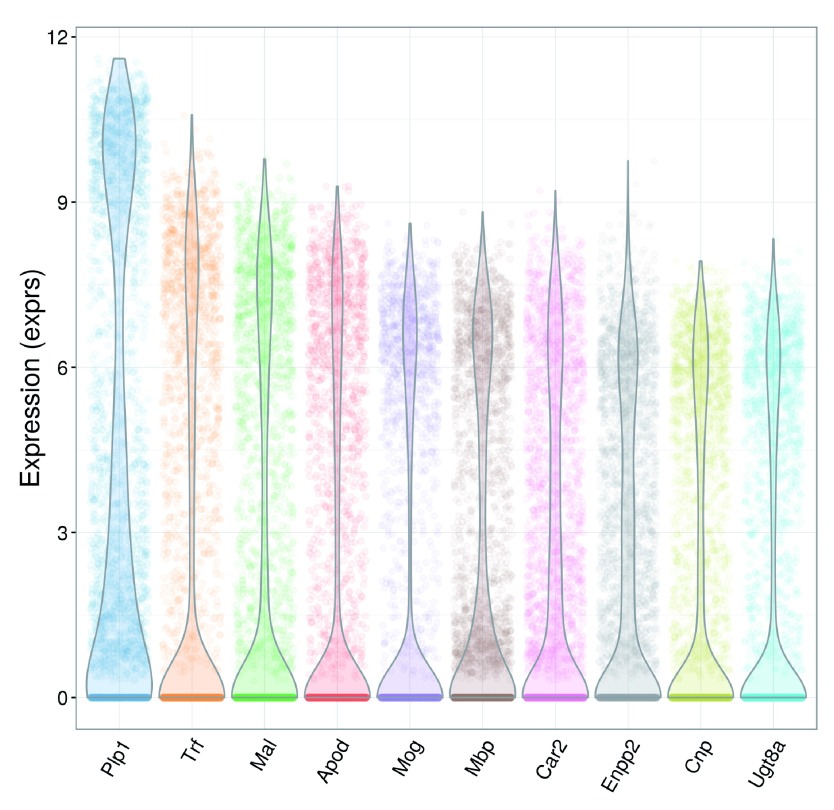
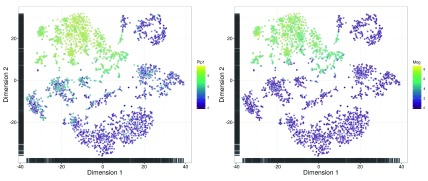
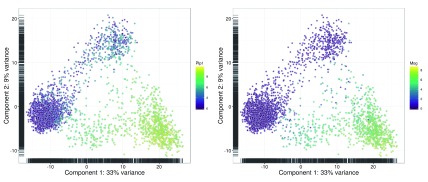
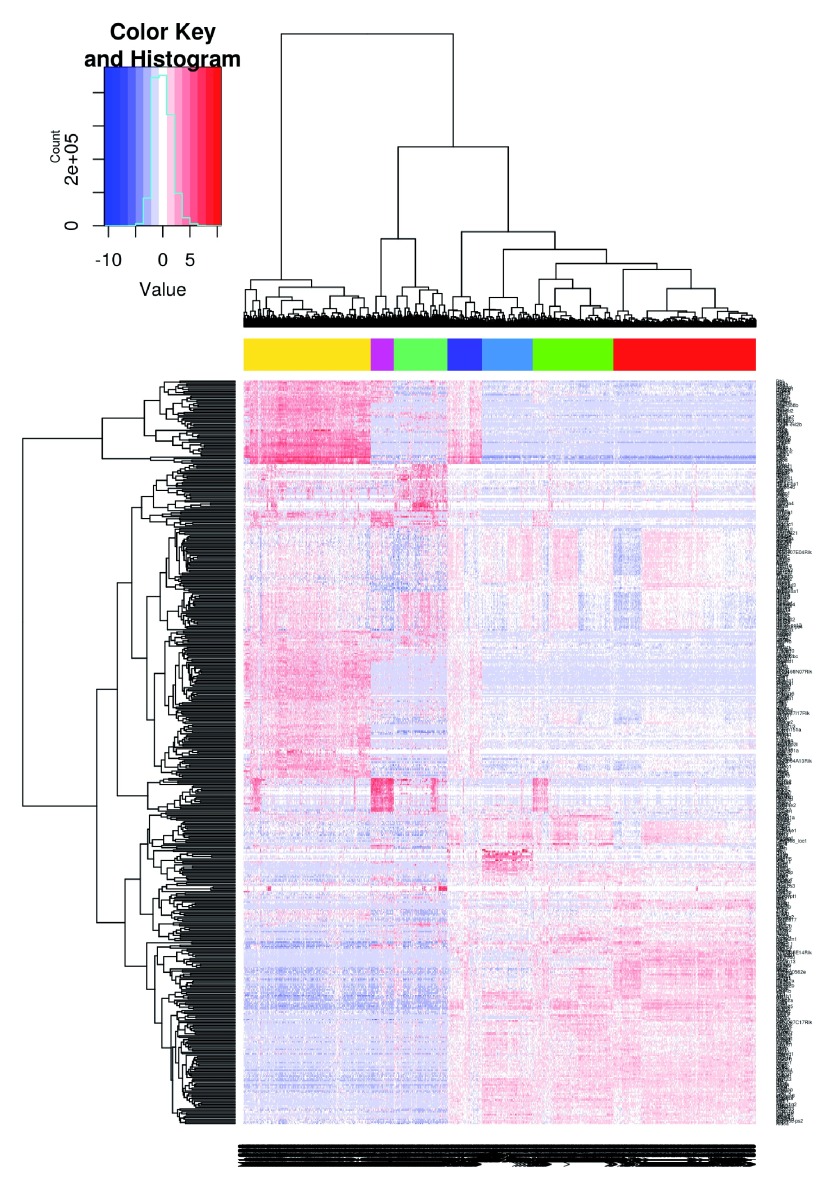
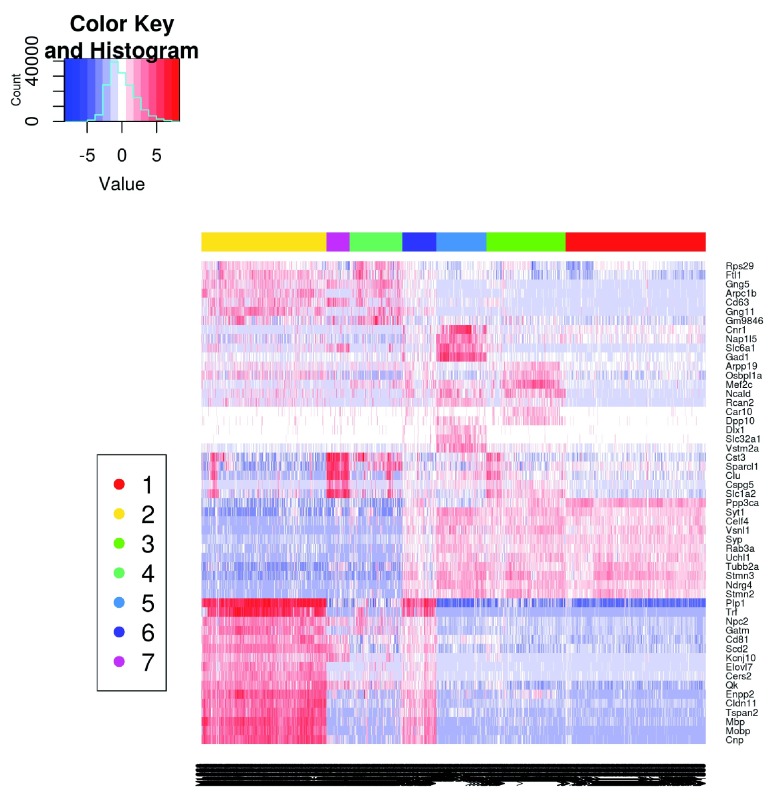
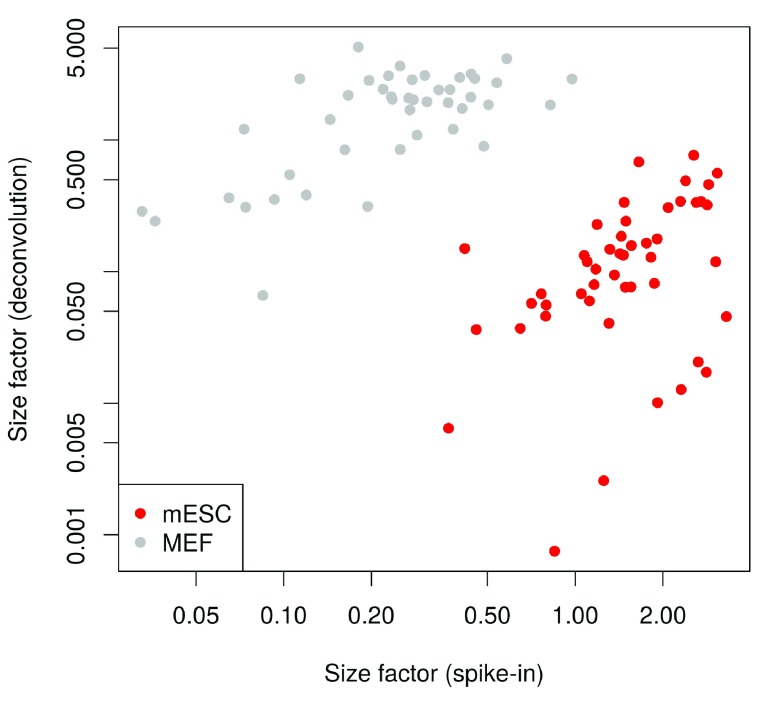
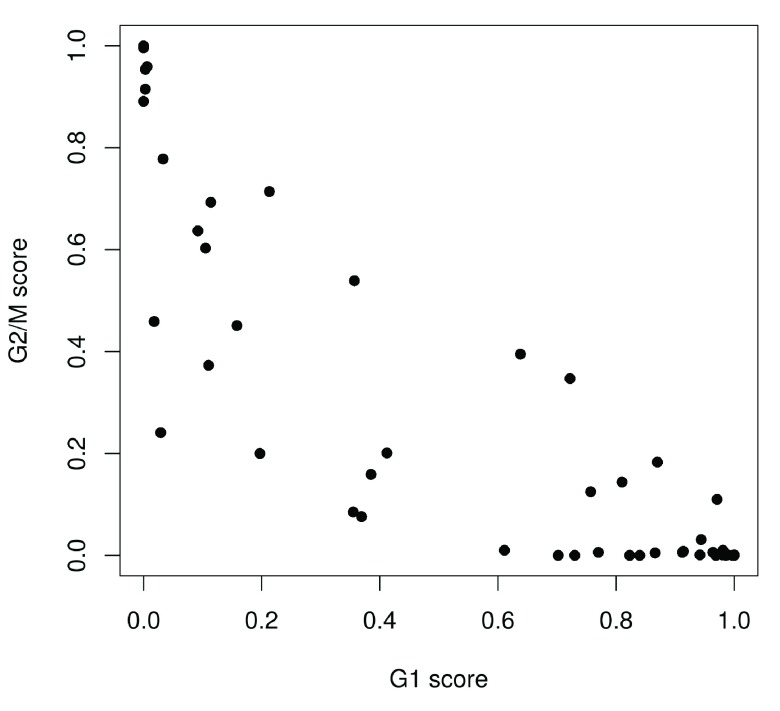
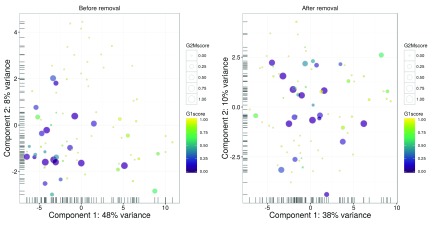
Single-cell RNA sequencing (scRNA-seq) is widely used to profile the transcriptome of individual cells. This provides biological resolution that cannot be matched by bulk RNA sequencing, at the cost of increased technical noise and data complexity. The differences between scRNA-seq and bulk RNA-seq data mean that the analysis of the former cannot be performed by recycling bioinformatics pipelines for the latter. Rather, dedicated single-cell methods are required at various steps to exploit the cellular resolution while accounting for technical noise. This article describes a computational workflow for low-level analyses of scRNA-seq data, based primarily on software packages from the open-source Bioconductor project. It covers basic steps including quality control, data exploration and normalization, as well as more complex procedures such as cell cycle phase assignment, identification of highly variable and correlated genes, clustering into subpopulations and marker gene detection. Analyses were demonstrated on gene-level count data from several publicly available datasets involving haematopoietic stem cells, brain-derived cells, T-helper cells and mouse embryonic stem cells. This will provide a range of usage scenarios from which readers can construct their own analysis pipelines.
单细胞RNA测序(scRNA-seq)被广泛用于分析单个细胞的转录组。这提供了整体RNA测序无法比拟的生物学分辨率,但代价是技术噪声增加和数据复杂性提高。scRNA-seq数据与整体RNA-seq数据的差异意味着前者的分析不能通过复用后者的生物信息学流程来进行。相反,在各个步骤都需要专门的单细胞方法,以利用细胞分辨率,同时考虑技术噪声。本文介绍了一种基于开源Bioconductor项目的软件包进行scRNA-seq数据低级分析的计算工作流程。它涵盖了基本步骤,包括质量控制、数据探索和标准化,以及更复杂的程序,如细胞周期阶段分配、高变和相关基因的识别、聚类成亚群和标记基因检测。对来自几个公开可用数据集(涉及造血干细胞、脑源细胞、辅助性T细胞和小鼠胚胎干细胞)的基因水平计数数据进行了分析演示。这将提供一系列使用场景,读者可以从中构建自己的分析流程。