Edge Peter, Bafna Vineet, Bansal Vikas
Department of Computer Science & Engineering, University of California, San Diego, La Jolla, California 92053, USA.
Department of Pediatrics, School of Medicine, University of California, San Diego, La Jolla, California 92053, USA.
Genome Res. 2017 May;27(5):801-812. doi: 10.1101/gr.213462.116. Epub 2016 Dec 9.
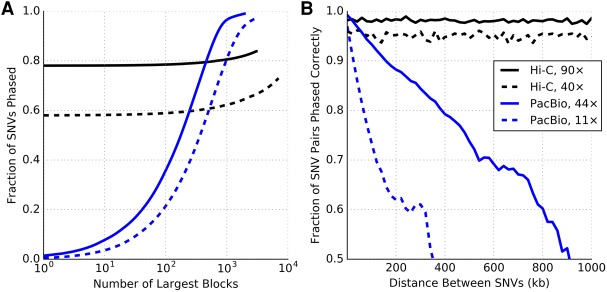
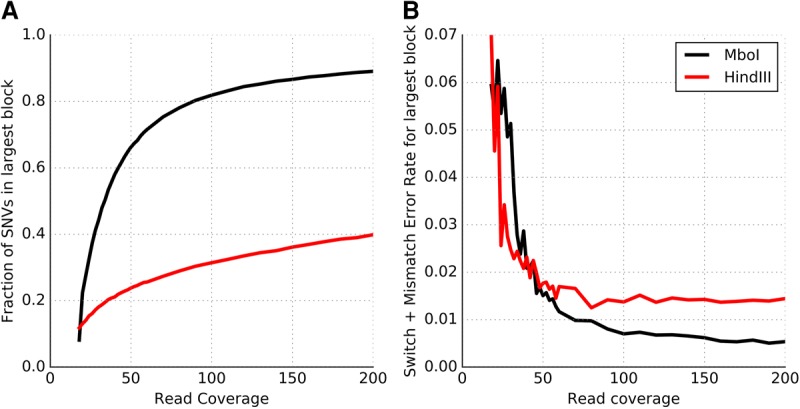
Many tools have been developed for haplotype assembly-the reconstruction of individual haplotypes using reads mapped to a reference genome sequence. Due to increasing interest in obtaining haplotype-resolved human genomes, a range of new sequencing protocols and technologies have been developed to enable the reconstruction of whole-genome haplotypes. However, existing computational methods designed to handle specific technologies do not scale well on data from different protocols. We describe a new algorithm, HapCUT2, that extends our previous method (HapCUT) to handle multiple sequencing technologies. Using simulations and whole-genome sequencing (WGS) data from multiple different data types-dilution pool sequencing, linked-read sequencing, single molecule real-time (SMRT) sequencing, and proximity ligation (Hi-C) sequencing-we show that HapCUT2 rapidly assembles haplotypes with best-in-class accuracy for all data types. In particular, HapCUT2 scales well for high sequencing coverage and rapidly assembled haplotypes for two long-read WGS data sets on which other methods struggled. Further, HapCUT2 directly models Hi-C specific error modalities, resulting in significant improvements in error rates compared to HapCUT, the only other method that could assemble haplotypes from Hi-C data. Using HapCUT2, haplotype assembly from a 90× coverage whole-genome Hi-C data set yielded high-resolution haplotypes (78.6% of variants phased in a single block) with high pairwise phasing accuracy (∼98% across chromosomes). Our results demonstrate that HapCUT2 is a robust tool for haplotype assembly applicable to data from diverse sequencing technologies.
已经开发了许多用于单倍型组装的工具,即利用映射到参考基因组序列的 reads 来重建个体单倍型。由于对获得单倍型解析的人类基因组的兴趣日益增加,已经开发了一系列新的测序方案和技术,以实现全基因组单倍型的重建。然而,现有的旨在处理特定技术的计算方法在处理来自不同方案的数据时扩展性不佳。我们描述了一种新算法 HapCUT2,它扩展了我们之前的方法(HapCUT)以处理多种测序技术。使用来自多种不同数据类型的模拟和全基因组测序(WGS)数据——稀释池测序、连接 reads 测序、单分子实时(SMRT)测序和邻近连接(Hi-C)测序——我们表明 HapCUT2 能快速组装单倍型,对所有数据类型都具有一流的准确性。特别是,HapCUT2 在高测序覆盖度下扩展性良好,并能快速为另外两种其他方法难以处理的长 reads WGS 数据集组装单倍型。此外,HapCUT2 直接对 Hi-C 特定的错误模式进行建模,与唯一一种也能从 Hi-C 数据组装单倍型的其他方法 HapCUT 相比,错误率有显著改善。使用 HapCUT2,从 90×覆盖度的全基因组 Hi-C 数据集组装单倍型产生了高分辨率单倍型(78.6%的变异在单个区域内被定相),且具有高的成对定相准确性(跨染色体约 98%)。我们的结果表明,HapCUT2 是一种适用于来自多种测序技术数据的稳健的单倍型组装工具。