Zheng Grace X Y, Lau Billy T, Schnall-Levin Michael, Jarosz Mirna, Bell John M, Hindson Christopher M, Kyriazopoulou-Panagiotopoulou Sofia, Masquelier Donald A, Merrill Landon, Terry Jessica M, Mudivarti Patrice A, Wyatt Paul W, Bharadwaj Rajiv, Makarewicz Anthony J, Li Yuan, Belgrader Phillip, Price Andrew D, Lowe Adam J, Marks Patrick, Vurens Gerard M, Hardenbol Paul, Montesclaros Luz, Luo Melissa, Greenfield Lawrence, Wong Alexander, Birch David E, Short Steven W, Bjornson Keith P, Patel Pranav, Hopmans Erik S, Wood Christina, Kaur Sukhvinder, Lockwood Glenn K, Stafford David, Delaney Joshua P, Wu Indira, Ordonez Heather S, Grimes Susan M, Greer Stephanie, Lee Josephine Y, Belhocine Kamila, Giorda Kristina M, Heaton William H, McDermott Geoffrey P, Bent Zachary W, Meschi Francesca, Kondov Nikola O, Wilson Ryan, Bernate Jorge A, Gauby Shawn, Kindwall Alex, Bermejo Clara, Fehr Adrian N, Chan Adrian, Saxonov Serge, Ness Kevin D, Hindson Benjamin J, Ji Hanlee P
10X Genomics, Pleasanton, California, USA.
Stanford Genome Technology Center, Stanford University, Palo Alto, California, USA.
Nat Biotechnol. 2016 Mar;34(3):303-11. doi: 10.1038/nbt.3432. Epub 2016 Feb 1.
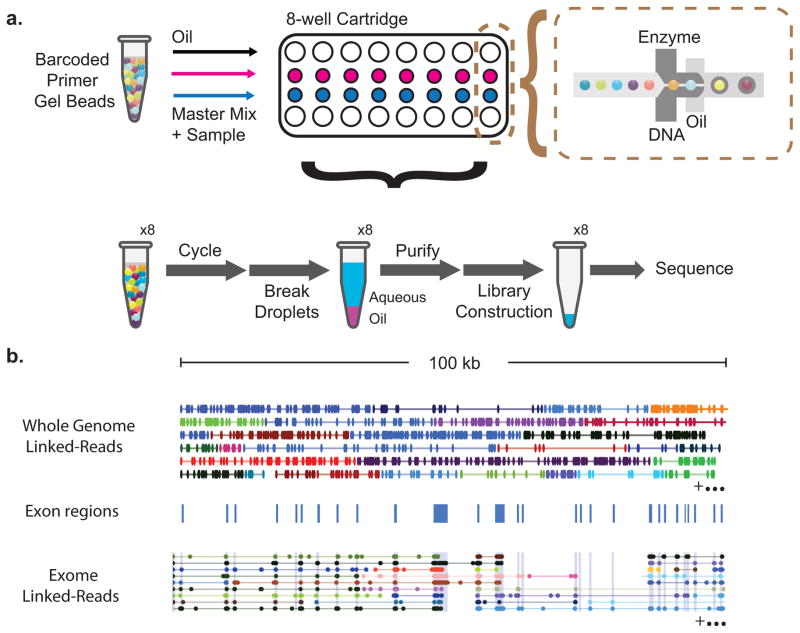
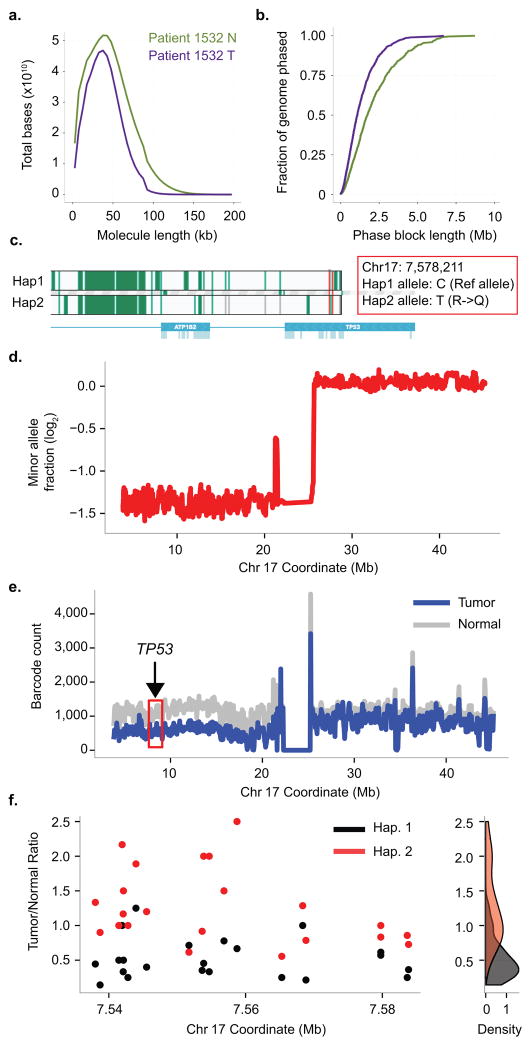
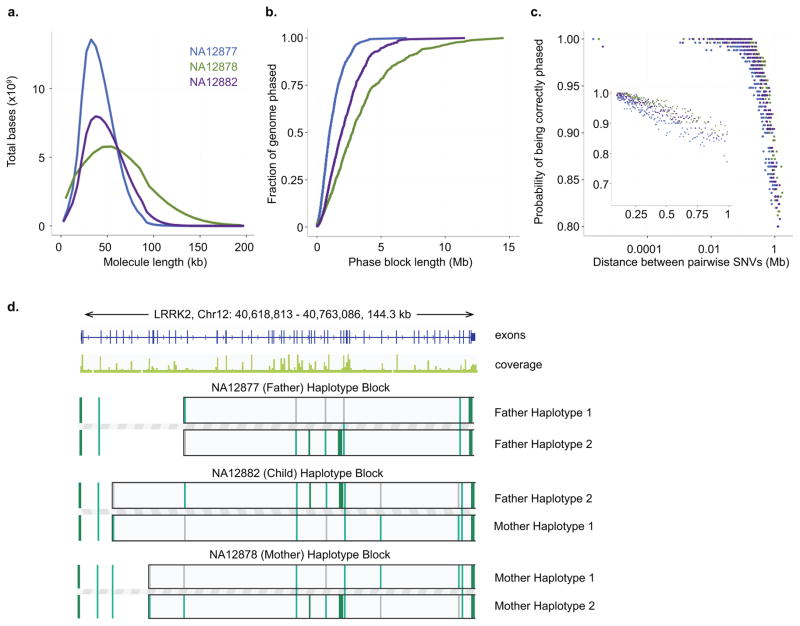
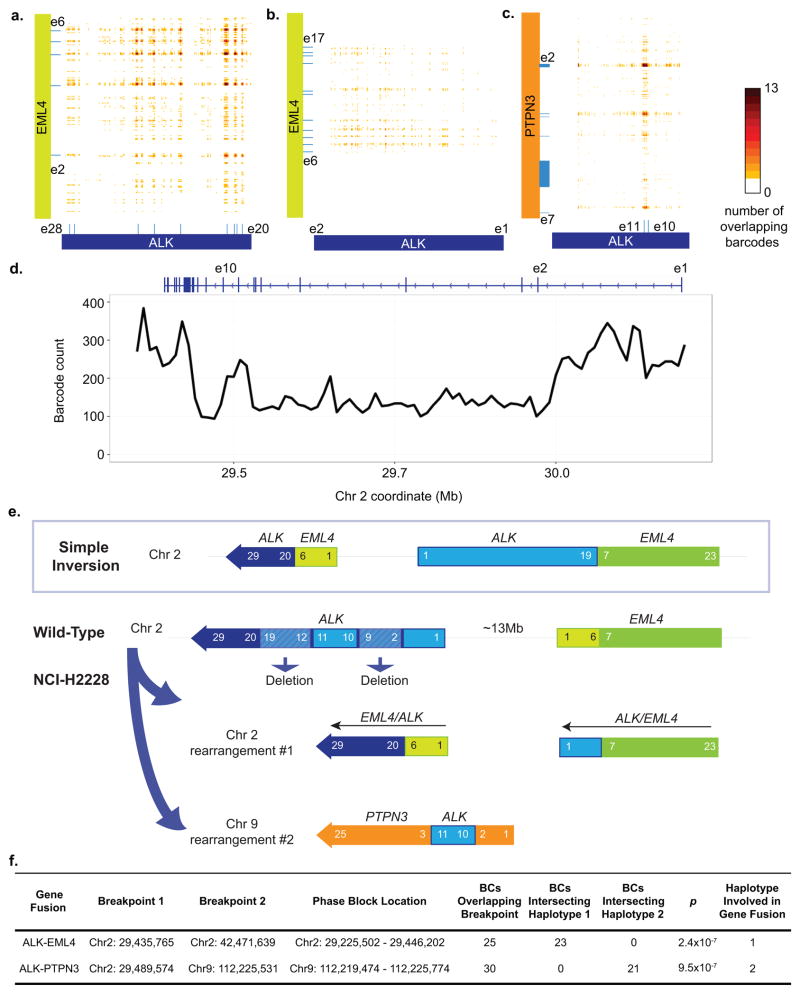
Haplotyping of human chromosomes is a prerequisite for cataloguing the full repertoire of genetic variation. We present a microfluidics-based, linked-read sequencing technology that can phase and haplotype germline and cancer genomes using nanograms of input DNA. This high-throughput platform prepares barcoded libraries for short-read sequencing and computationally reconstructs long-range haplotype and structural variant information. We generate haplotype blocks in a nuclear trio that are concordant with expected inheritance patterns and phase a set of structural variants. We also resolve the structure of the EML4-ALK gene fusion in the NCI-H2228 cancer cell line using phased exome sequencing. Finally, we assign genetic aberrations to specific megabase-scale haplotypes generated from whole-genome sequencing of a primary colorectal adenocarcinoma. This approach resolves haplotype information using up to 100 times less genomic DNA than some methods and enables the accurate detection of structural variants.
对人类染色体进行单倍型分型是编目遗传变异完整清单的前提条件。我们提出了一种基于微流控的连锁读长测序技术,该技术能够使用纳克级的输入DNA对种系和癌症基因组进行定相和单倍型分型。这个高通量平台为短读长测序制备带条形码的文库,并通过计算重建长程单倍型和结构变异信息。我们在一个核心三人组中生成了与预期遗传模式一致的单倍型块,并对一组结构变异进行了定相。我们还使用定相外显子组测序解析了NCI-H2228癌细胞系中EML4-ALK基因融合的结构。最后,我们将遗传畸变定位到从原发性结肠直肠腺癌全基因组测序产生的特定兆碱基规模单倍型上。这种方法使用的基因组DNA比某些方法少多达100倍,就能解析单倍型信息,并能准确检测结构变异。