Amini Sasan, Pushkarev Dmitry, Christiansen Lena, Kostem Emrah, Royce Tom, Turk Casey, Pignatelli Natasha, Adey Andrew, Kitzman Jacob O, Vijayan Kandaswamy, Ronaghi Mostafa, Shendure Jay, Gunderson Kevin L, Steemers Frank J
Illumina, Inc., Advanced Research Group, San Diego, California, USA.
Department of Genome Sciences, University of Washington, Seattle, Washington, USA.
Nat Genet. 2014 Dec;46(12):1343-9. doi: 10.1038/ng.3119. Epub 2014 Oct 19.
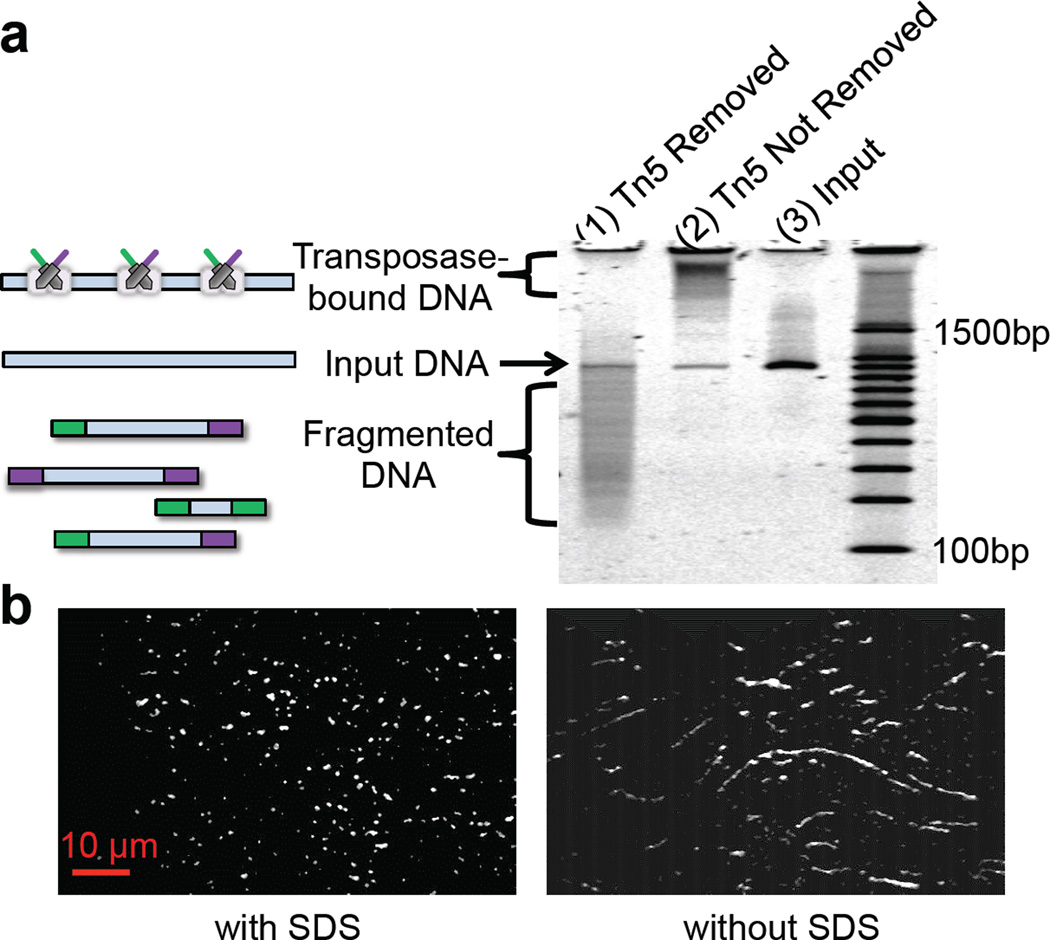
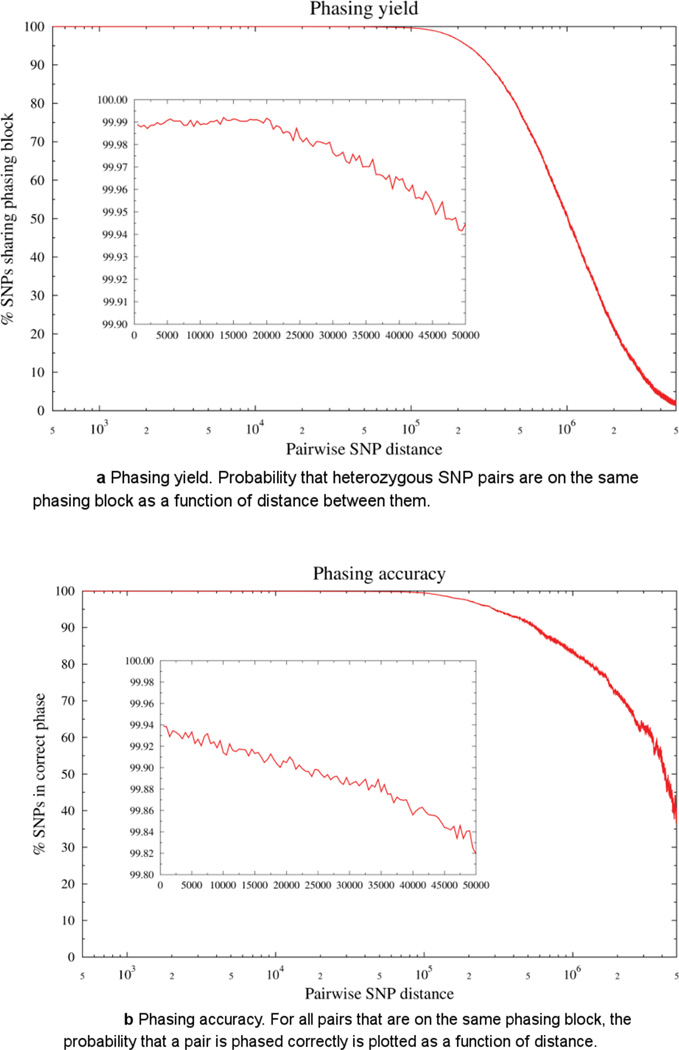
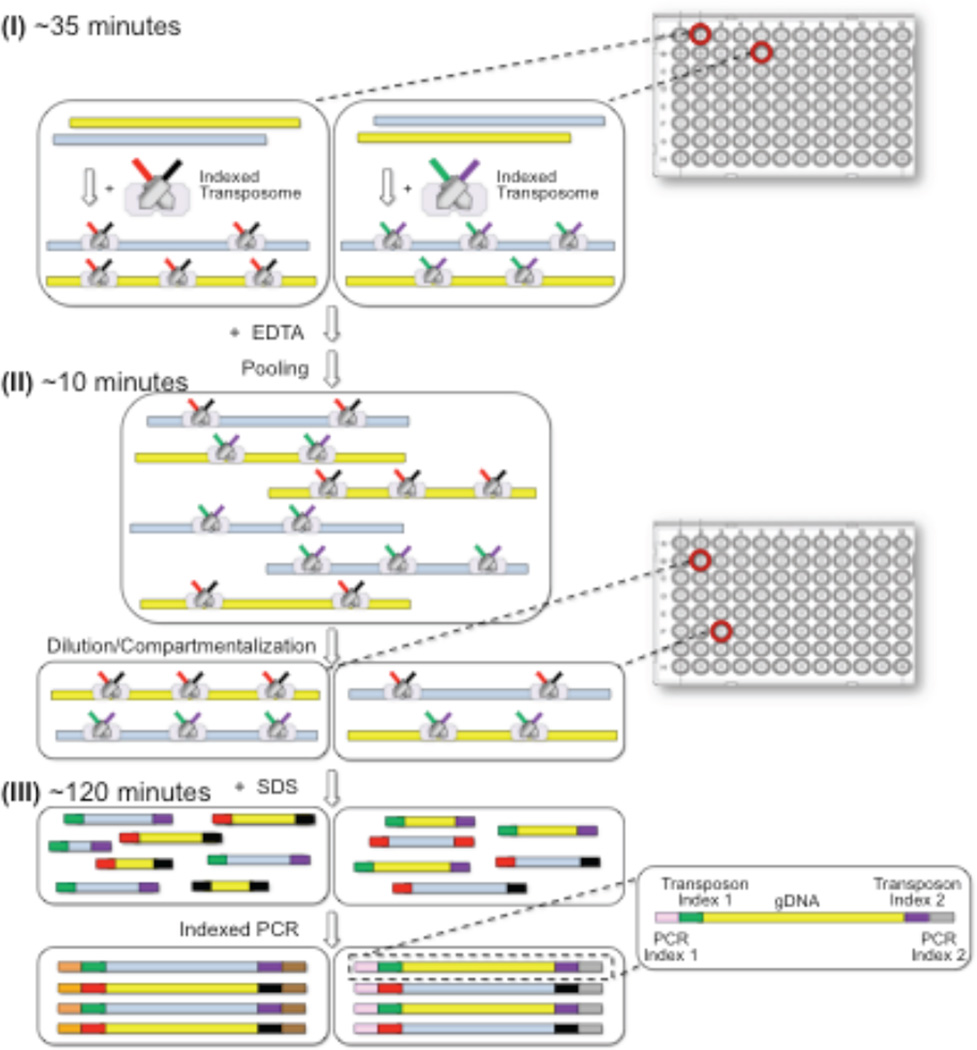
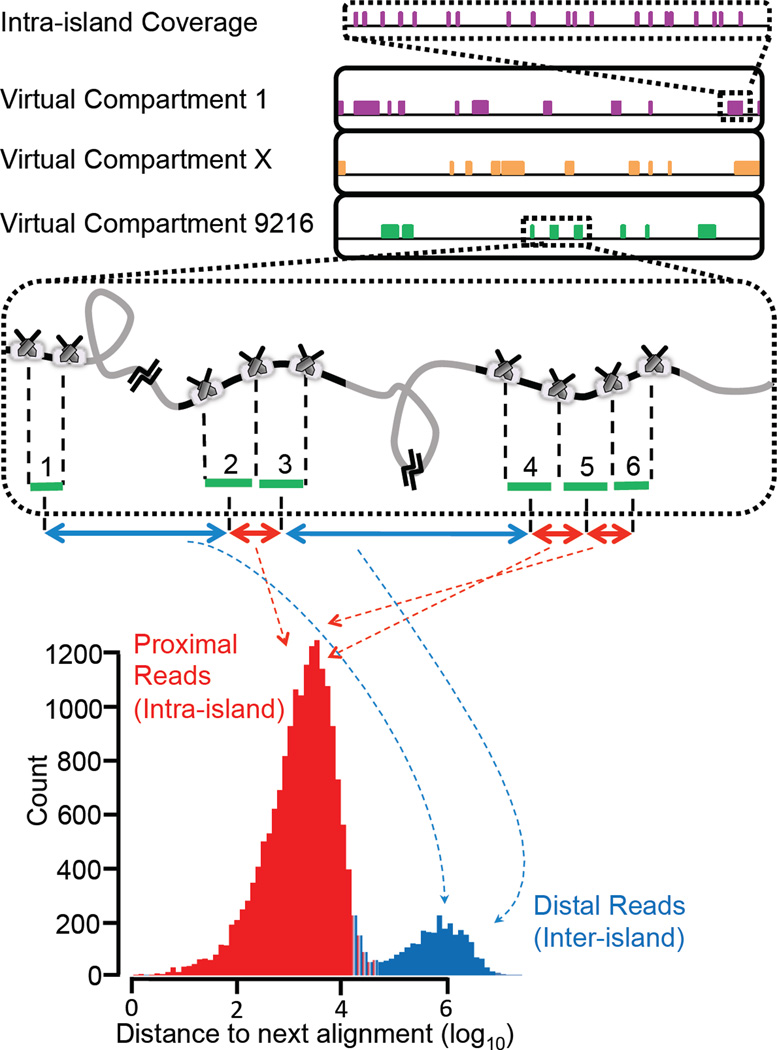
Haplotype-resolved genome sequencing enables the accurate interpretation of medically relevant genetic variation, deep inferences regarding population history and non-invasive prediction of fetal genomes. We describe an approach for genome-wide haplotyping based on contiguity-preserving transposition (CPT-seq) and combinatorial indexing. Tn5 transposition is used to modify DNA with adaptor and index sequences while preserving contiguity. After DNA dilution and compartmentalization, the transposase is removed, resolving the DNA into individually indexed libraries. The libraries in each compartment, enriched for neighboring genomic elements, are further indexed via PCR. Combinatorial 96-plex indexing at both the transposition and PCR stage enables the construction of phased synthetic reads from each of the nearly 10,000 'virtual compartments'. We demonstrate the feasibility of this method by assembling >95% of the heterozygous variants in a human genome into long, accurate haplotype blocks (N50 = 1.4-2.3 Mb). The rapid, scalable and cost-effective workflow could enable haplotype resolution to become routine in human genome sequencing.
单倍型解析基因组测序能够准确解读医学相关的基因变异,深入推断群体历史,并对胎儿基因组进行无创预测。我们描述了一种基于保序转座(CPT-seq)和组合索引的全基因组单倍型分型方法。Tn5转座用于用衔接子和索引序列修饰DNA,同时保持连续性。在DNA稀释和分区后,去除转座酶,将DNA解析为单独索引的文库。每个分区中富含相邻基因组元件的文库通过PCR进一步索引。在转座和PCR阶段的组合96重索引能够从近10000个“虚拟分区”中的每一个构建分阶段的合成读数。我们通过将人类基因组中>95%的杂合变异组装成长而准确的单倍型块(N50 = 1.4 - 2.3 Mb)来证明该方法的可行性。这种快速、可扩展且经济高效的工作流程可使单倍型解析在人类基因组测序中成为常规操作。