Shah Ruth R, Cholewa-Waclaw Justyna, Davies Faith C J, Paton Katie M, Chaligne Ronan, Heard Edith, Abbott Catherine M, Bird Adrian P
Wellcome Trust Centre for Cell Biology, University of Edinburgh, Edinburgh, UK.
Centre for Genomic and Experimental Medicine, MRC Institute of Genetics and Molecular Medicine, University of Edinburgh, Western General Hospital, Edinburgh, UK.
Wellcome Open Res. 2016 Nov 15;1:13. doi: 10.12688/wellcomeopenres.10011.1.
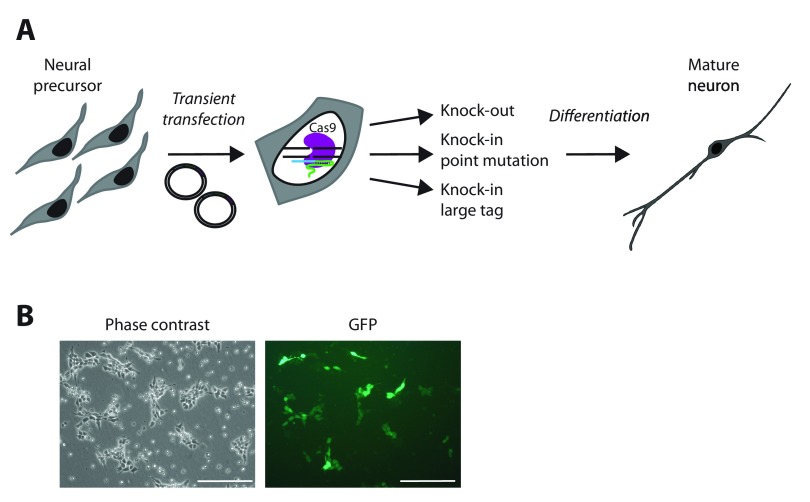
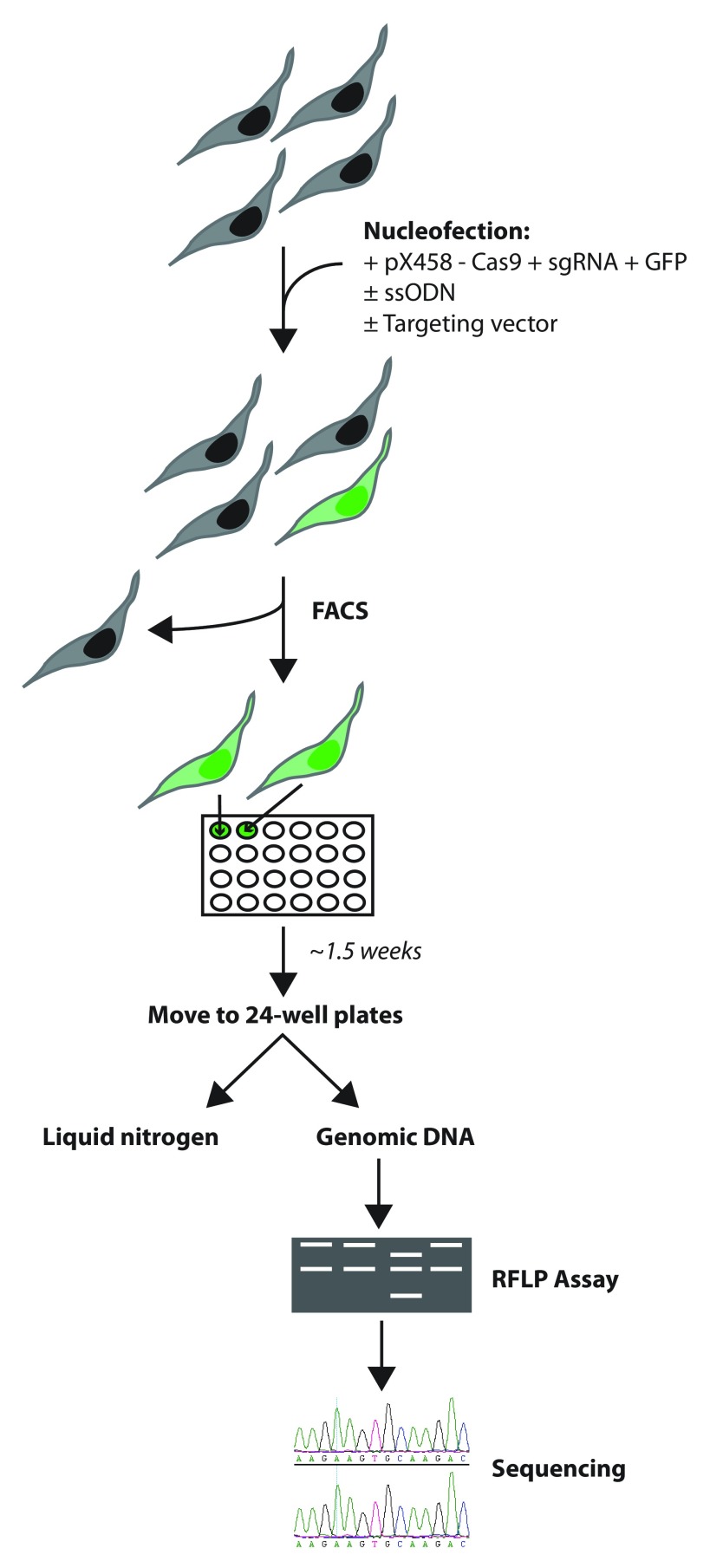
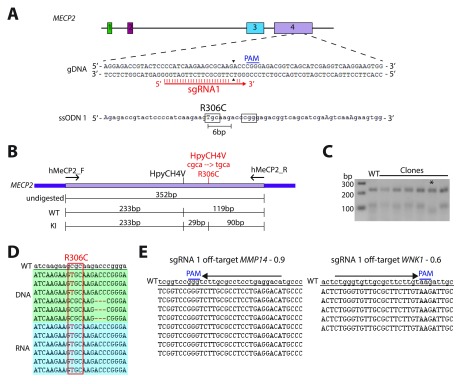
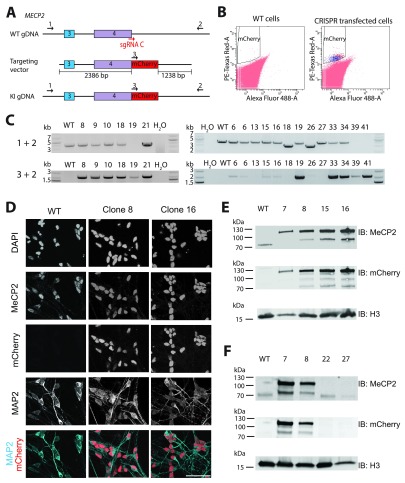
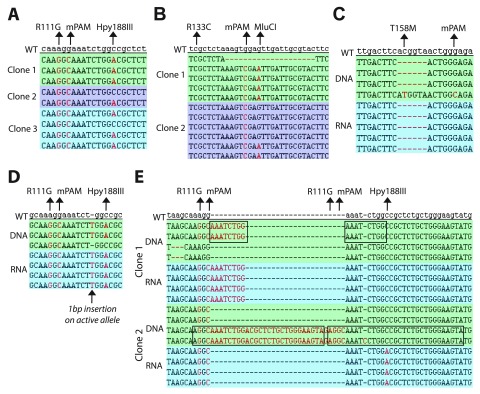
The recent identification of multiple new genetic causes of neurological disorders highlights the need for model systems that give experimental access to the underlying biology. In particular, the ability to couple disease-causing mutations with human neuronal differentiation systems would be beneficial. Gene targeting is a well-known approach for dissecting gene function, but low rates of homologous recombination in somatic cells (including neuronal cells) have traditionally impeded the development of robust cellular models of neurological disorders. Recently, however, CRISPR/Cas9 gene editing technologies have expanded the number of systems within which gene targeting is possible. Here we adopt as a model system LUHMES cells, a commercially available diploid human female mesencephalic cell line that differentiates into homogeneous mature neurons in 1-2 weeks. We describe optimised methods for transfection and selection of neuronal progenitor cells carrying targeted genomic alterations using CRISPR/Cas9 technology. By targeting the endogenous X-linked locus, we introduced four independent missense mutations that cause the autism spectrum disorder Rett syndrome and observed the desired genetic structure in 3-26% of selected clones, including gene targeting of the inactive X chromosome. Similar efficiencies were achieved by introducing neurodevelopmental disorder-causing mutations at the autosomal locus on chromosome 20. Our results indicate that efficiency of genetic "knock-in" is determined by the location of the mutation within the donor DNA molecule. Furthermore, we successfully introduced an mCherry tag at the locus to yield a fusion protein, demonstrating that larger insertions are also straightforward in this system. We suggest that our optimised methods for altering the genome of LUHMES cells make them an attractive model for the study of neurogenetic disorders.
最近对多种神经系统疾病新遗传病因的鉴定凸显了建立能够对潜在生物学机制进行实验研究的模型系统的必要性。特别是,将致病突变与人类神经元分化系统相结合的能力将大有裨益。基因靶向是一种众所周知的剖析基因功能的方法,但体细胞(包括神经元细胞)中同源重组率低传统上阻碍了强大的神经系统疾病细胞模型的开发。然而,最近CRISPR/Cas9基因编辑技术扩大了可以进行基因靶向的系统数量。在这里,我们采用LUHMES细胞作为模型系统,它是一种市售的二倍体人类雌性中脑细胞系,可在1-2周内分化为同质的成熟神经元。我们描述了使用CRISPR/Cas9技术转染和筛选携带靶向基因组改变的神经元祖细胞的优化方法。通过靶向内源性X连锁基因座,我们引入了四个独立的错义突变,这些突变导致自闭症谱系障碍雷特综合征,并在3-26%的选定克隆中观察到了所需的遗传结构,包括对失活X染色体的基因靶向。通过在20号染色体的常染色体基因座上引入导致神经发育障碍的突变,也实现了类似的效率。我们的结果表明,基因“敲入”的效率取决于供体DNA分子内突变的位置。此外,我们成功地在该基因座引入了一个mCherry标签以产生融合蛋白,这表明在该系统中进行更大的插入也很简单。我们认为,我们优化的改变LUHMES细胞基因组的方法使其成为研究神经遗传疾病具有吸引力的模型。