Madrigal Pedro
Wellcome Trust-Medical Research Council Cambridge Stem Cell Institute, University of Cambridge, Cambridge CB2 0SZ, UK.
Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hixton CB10 1SA, UK.
Bioinformatics. 2017 Mar 1;33(5):746-748. doi: 10.1093/bioinformatics/btw724.
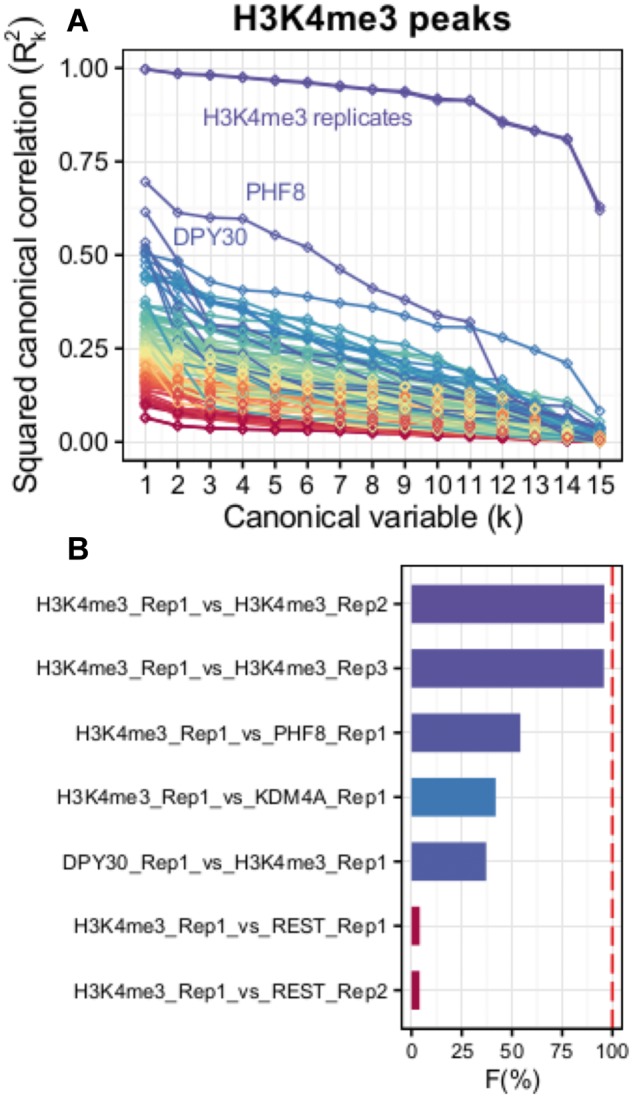
Computational evaluation of variability across DNA or RNA sequencing datasets is a crucial step in genomic science, as it allows both to evaluate reproducibility of biological or technical replicates, and to compare different datasets to identify their potential correlations. Here we present fCCAC, an application of functional canonical correlation analysis to assess covariance of nucleic acid sequencing datasets such as chromatin immunoprecipitation followed by deep sequencing (ChIP-seq). We show how this method differs from other measures of correlation, and exemplify how it can reveal shared covariance between histone modifications and DNA binding proteins, such as the relationship between the H3K4me3 chromatin mark and its epigenetic writers and readers.
An R/Bioconductor package is available at http://bioconductor.org/packages/fCCAC/ .
Supplementary data are available at Bioinformatics online.
对DNA或RNA测序数据集的变异性进行计算评估是基因组科学中的关键步骤,因为它既能评估生物学或技术重复的可重复性,又能比较不同数据集以识别它们潜在的相关性。在此,我们展示了fCCAC,这是一种功能典型相关分析的应用,用于评估核酸测序数据集的协方差,如染色质免疫沉淀测序(ChIP-seq)。我们展示了该方法与其他相关性度量方法的不同之处,并举例说明了它如何揭示组蛋白修饰与DNA结合蛋白之间的共享协方差,例如H3K4me3染色质标记与其表观遗传书写器和读取器之间的关系。
可在http://bioconductor.org/packages/fCCAC/获取R/Bioconductor软件包。
补充数据可在《生物信息学》在线获取。