Brown Scott D, Hapgood Greg, Steidl Christian, Weng Andrew P, Savage Kerry J, Holt Robert A
Canada's Michael Smith Genome Sciences Centre, BC Cancer Agency, Vancouver, British Columbia V5Z 1L3, Canada.
Genome Science and Technology Program, University of British Columbia, Vancouver, British Columbia V6T 1Z4, Canada.
Bioinformatics. 2017 Apr 15;33(8):1111-1115. doi: 10.1093/bioinformatics/btw810.
In T-cell lymphoma, malignant T cells arising from a founding clone share an identical T cell receptor (TCR) and can be identified by the over-representation of this TCR relative to TCRs from the patient's repertoire of normal T cells. Here, we demonstrate that TCR information extracted from RNA-seq data can provide a higher resolution view of peripheral T cell lymphomas (PTCLs) than that provided by conventional methods.
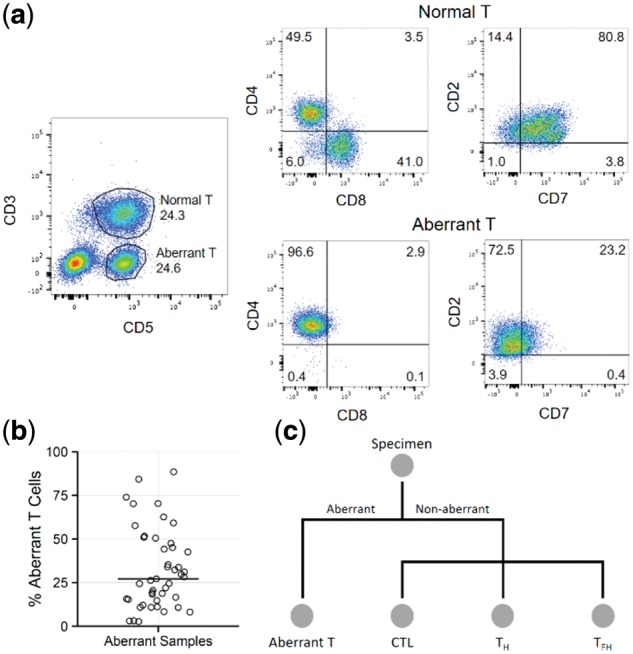
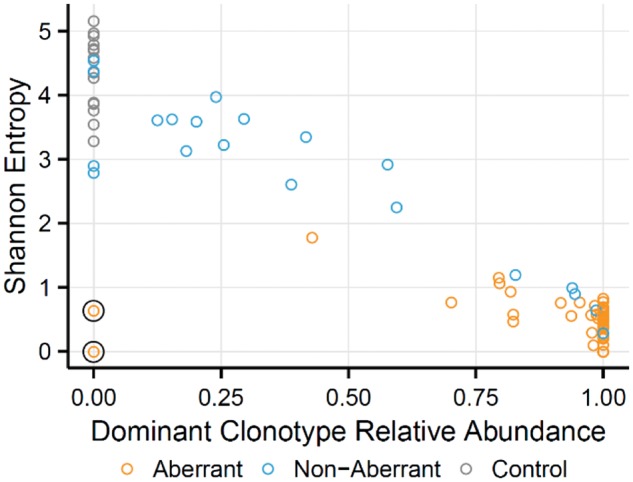
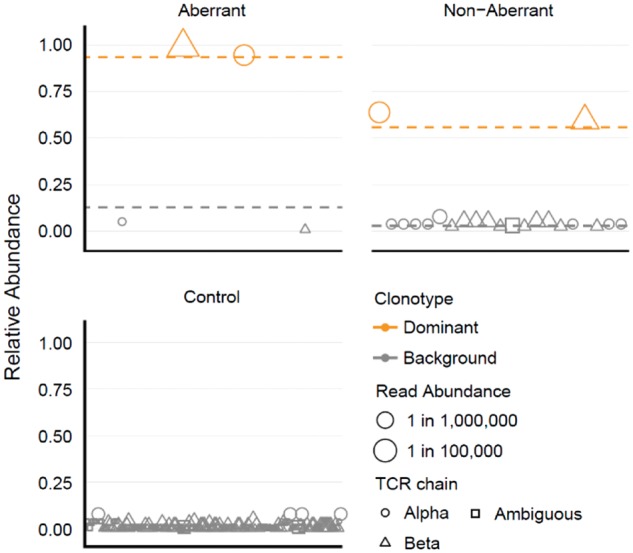
For 60 subjects with PTCL, flow cytometry/FACS was used to identify and sort aberrant T cell populations from diagnostic lymph node cell suspensions. For samples that did not appear to contain aberrant T cell populations, T helper (T H ), T follicular helper (T FH ) and cytotoxic T lymphocyte (CTL) subsets were sorted. RNA-seq was performed on sorted T cell populations, and TCR alpha and beta chain sequences were extracted and quantified directly from the RNA-seq data. 96% of the immunophenotypically aberrant samples had a dominant T cell clone readily identifiable by RNA-seq. Of the samples where no aberrant population was found by flow cytometry, 80% had a dominant clone by RNA-seq. This demonstrates the increased sensitivity and diagnostic ability of RNA-seq over flow cytometry and shows that the presence of a normal immunophenotype does not exclude clonality.
R scripts used in the processing of the data are available online at https://www.github.com/scottdbrown/RNAseq-TcellClonality.
rholt@bcgsc.ca or ksavage@bccancer.bc.ca.
Supplementary data are available at Bioinformatics online.
在T细胞淋巴瘤中,源自一个起始克隆的恶性T细胞共享相同的T细胞受体(TCR),并且可以通过该TCR相对于患者正常T细胞库中的TCR的过度表达来识别。在这里,我们证明从RNA测序数据中提取的TCR信息可以提供比传统方法更高分辨率的外周T细胞淋巴瘤(PTCL)视图。
对于60例PTCL患者,使用流式细胞术/荧光激活细胞分选术从诊断性淋巴结细胞悬液中鉴定和分选异常T细胞群体。对于似乎不包含异常T细胞群体的样本,分选辅助性T细胞(TH)、滤泡辅助性T细胞(TFH)和细胞毒性T淋巴细胞(CTL)亚群。对分选的T细胞群体进行RNA测序,并直接从RNA测序数据中提取和定量TCRα和β链序列。96%的免疫表型异常样本有一个通过RNA测序易于识别的优势T细胞克隆。在流式细胞术未发现异常群体的样本中,80%通过RNA测序有一个优势克隆。这证明了RNA测序相对于流式细胞术的敏感性和诊断能力增强,并且表明正常免疫表型的存在并不排除克隆性。
处理数据中使用的R脚本可在https://www.github.com/scottdbrown/RNAseq-TcellClonality在线获取。
rholt@bcgsc.ca或ksavage@bccancer.bc.ca。
补充数据可在《生物信息学》在线获取。