Luzzago Camilla, Ebranati Erika, Cabezón Oscar, Fernández-Sirera Laura, Lavín Santiago, Rosell Rosa, Veo Carla, Rossi Luca, Cavallero Serena, Lanfranchi Paolo, Marco Ignasi, Zehender Gianguglielmo
Department of Veterinary Medicine, University of Milan, Milano, Italy.
Centro di Ricerca Coordinata Epidemiologia e Sorveglianza Molecolare delle Infezioni-EpiSoMI, University of Milan, Milano, Italy.
PLoS One. 2016 Dec 29;11(12):e0168232. doi: 10.1371/journal.pone.0168232. eCollection 2016.
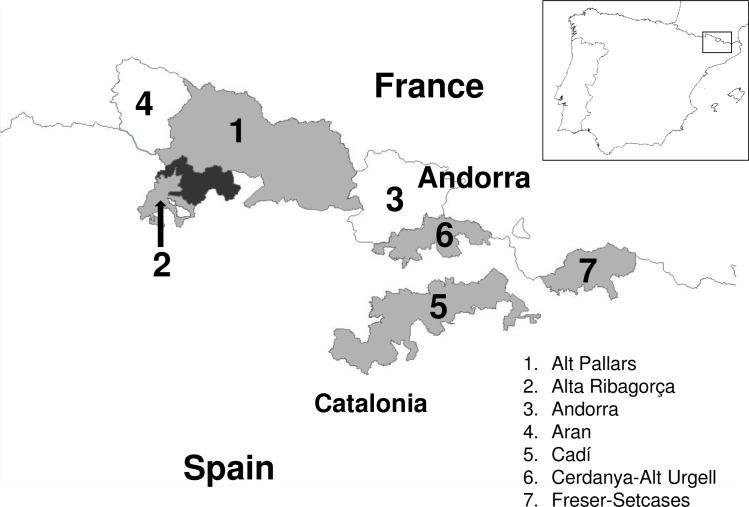
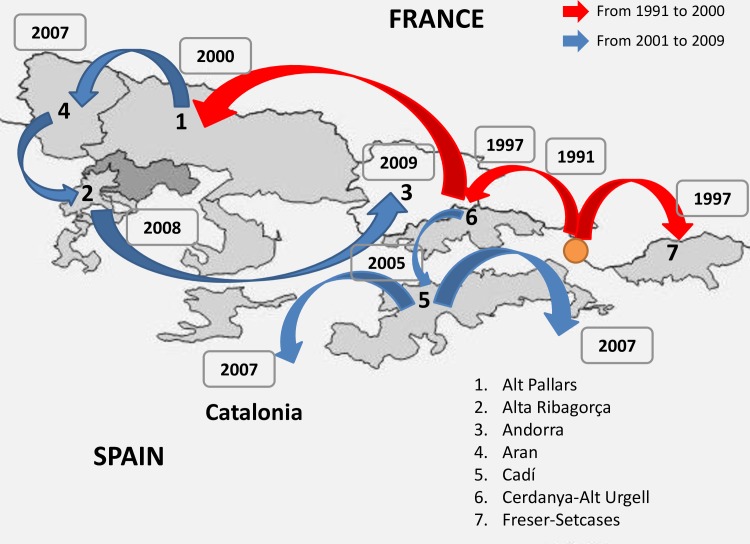
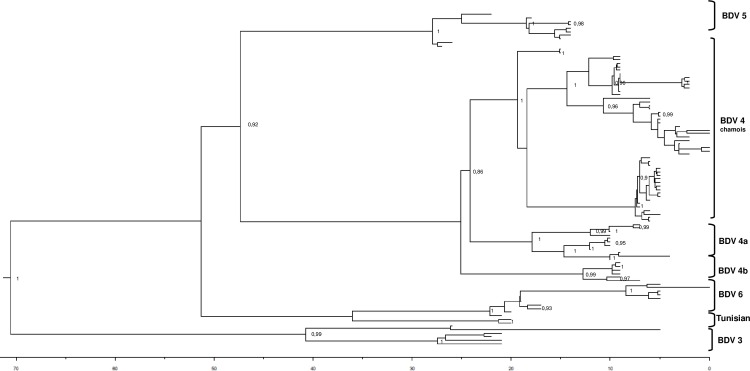
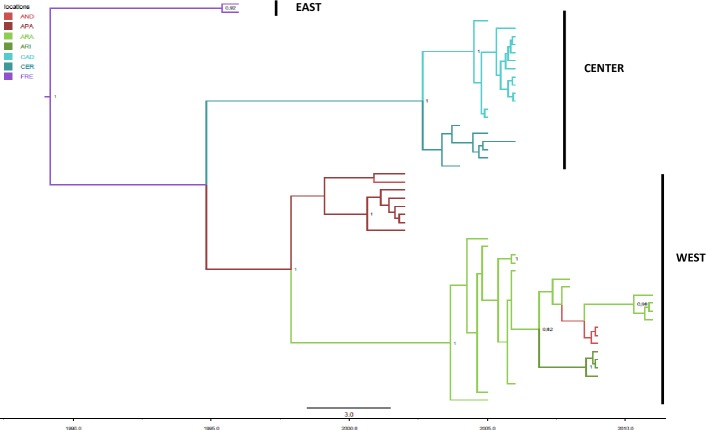
Border disease virus (BDV) affects a wide range of ruminants worldwide, mainly domestic sheep and goat. Since 2001 several outbreaks of disease associated to BDV infection have been described in Pyrenean chamois (Rupicapra pyrenaica pyrenaica) in Spain, France and Andorra. In order to reconstruct the most probable places of origin and pathways of dispersion of BDV among Pyrenean chamois, a phylogenetic analysis of 95 BDV 5'untranslated sequences has been performed on chamois and domestic ungulates, including novel sequences and retrieved from public databases, using a Bayesian Markov Chain Monte Carlo method. Discrete and continuous space phylogeography have been applied on chamois sequences dataset, using centroid positions and latitude and longitude coordinates of the animals, respectively. The estimated mean evolutionary rate of BDV sequences was 2.9×10-3 subs/site/year (95% HPD: 1.5-4.6×10-3). All the Pyrenean chamois isolates clustered in a unique highly significant clade, that originated from BDV-4a ovine clade. The introduction from sheep (dated back to the early 90s) generated a founder effect on the chamois population and the most probable place of origin of Pyrenean chamois BDV was estimated at coordinates 42.42 N and 1.9 E. The pathways of virus dispersion showed two main routes: the first started on the early 90s of the past century with a westward direction and the second arise in Central Pyrenees. The virus spread westward for more than 125 km and southward for about 50km and the estimated epidemic diffusion rate was about 13.1 km/year (95% HPD 5.2-21.4 km/year). The strong spatial structure, with strains from a single locality segregating together in homogeneous groups, and the significant pathways of viral dispersion among the areas, allowed to reconstruct both events of infection in a single area and of migrations, occurring between neighboring areas.
边界病病毒(BDV)在全球范围内影响着多种反刍动物,主要是家养绵羊和山羊。自2001年以来,西班牙、法国和安道尔的比利牛斯羚羊(Rupicapra pyrenaica pyrenaica)中出现了几起与BDV感染相关的疾病暴发。为了重建BDV在比利牛斯羚羊中最可能的起源地和传播途径,利用贝叶斯马尔可夫链蒙特卡罗方法,对95个BDV 5'非翻译序列进行了系统发育分析,这些序列来自羚羊和家养有蹄类动物,包括新序列和从公共数据库中检索到的序列。离散和连续空间系统地理学分别应用于羚羊序列数据集,使用动物的质心位置以及纬度和经度坐标。BDV序列的估计平均进化速率为2.9×10-3个替换/位点/年(95%最高后验密度区间:1.5-4.6×10-3)。所有比利牛斯羚羊分离株聚集在一个独特的、高度显著的分支中,该分支起源于BDV-4a绵羊分支。绵羊的引入(可追溯到90年代初)对比利牛斯羚羊种群产生了奠基者效应,比利牛斯羚羊BDV最可能的起源地估计在北纬42.42度和东经1.9度。病毒传播途径显示出两条主要路线:第一条始于上世纪90年代初,向西传播;第二条出现在比利牛斯山脉中部。病毒向西传播超过125公里,向南传播约50公里,估计的疫情扩散速率约为13.1公里/年(95%最高后验密度区间5.2-21.4公里/年)。强大的空间结构,即来自单个地点的毒株在同质群体中聚集在一起,以及区域间病毒传播的显著途径,使得能够重建单个区域内的感染事件以及相邻区域之间发生的迁移事件。